
Abstract 抽象的
Emerging evidence suggests that signaling pathways can be spatially regulated to ensure rapid and efficient responses to dynamically changing local cues. Ferroptosis is a recently defined form of lipid peroxidation-driven cell death. Although the molecular mechanisms underlying ferroptosis are emerging, spatial aspects of its signaling remain largely unexplored. By analyzing a public database, we found that a mitochondrial chaperone protein, glucose-regulated protein 75 (GRP75), may have a previously undefined role in regulating ferroptosis. This was subsequently validated. Interestingly, under ferroptotic conditions, GRP75 translocated from mitochondria to mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) and the cytosol. Further mechanistic studies revealed a highly spatial regulation of GRP75-mediated antiferroptotic signaling. Under ferroptotic conditions, lipid peroxidation predominantly accumulated at the ER, which activated protein kinase A (PKA) in a cAMP-dependent manner. In particular, a signaling microdomain, the outer mitochondrial membrane protein A-kinase anchor protein 1 (AKAP1)-anchored PKA, phosphorylated GRP75 at S148 in MAMs. This caused GRP75 to be sequestered outside the mitochondria, where it competed with Nrf2 for Keap1 binding through a conserved high-affinity RGD-binding motif, ETGE. Nrf2 was then stabilized and activated, leading to the transcriptional activation of a panel of antiferroptotic genes. Blockade of the PKA/GRP75 axis dramatically increased the responses of cancer cells to ferroptosis both in vivo and in vitro. Our identification a localized signaling cascade involved in protecting cancer cells from ferroptosis broadens our understanding of cellular defense mechanisms against ferroptosis and also provides a new target axis (AKAP1/PKA/GRP75) to improve the responses of cancer cells to ferroptosis.
新的证据表明,信号通路可以在空间上进行调节,以确保对动态变化的局部线索做出快速有效的反应。铁死亡是最近定义的脂质过氧化驱动的细胞死亡的一种形式。尽管铁死亡背后的分子机制正在出现,但其信号传导的空间方面仍然很大程度上未被探索。通过分析公共数据库,我们发现线粒体伴侣蛋白,葡萄糖调节蛋白 75 (GRP75),可能在调节铁死亡中具有先前未明确的作用。随后这一说法得到了验证。有趣的是,在铁死亡条件下,GRP75 从线粒体转移到线粒体相关内质网 (ER) 膜 (MAM) 和细胞质。进一步的机制研究揭示了 GRP75 介导的抗铁死亡信号的高度空间调节。在铁死亡条件下,脂质过氧化主要积聚于内质网,从而以 cAMP 依赖性方式激活蛋白激酶 A (PKA)。特别是,信号微结构域,线粒体外膜蛋白 A 激酶锚定蛋白 1 (AKAP1) 锚定的 PKA,在 MAM 中的 S148 处磷酸化 GRP75。这导致 GRP75 被隔离在线粒体外,并通过保守的高亲和力 RGD 结合基序 ETGE 与 Nrf2 竞争 Keap1 的结合。然后 Nrf2 被稳定并激活,导致一组抗铁死亡基因的转录激活。阻断 PKA/GRP75 轴可显着增加体内和体外癌细胞对铁死亡的反应。 我们鉴定出参与保护癌细胞免遭铁死亡的局部信号级联,拓宽了我们对细胞铁死亡防御机制的理解,并提供了一个新的靶轴(AKAP1/PKA/GRP75)来改善癌细胞对铁死亡的反应。
Similar content being viewed by others
其他人正在查看类似内容


Introduction 介绍
Ferroptosis is a recently defined form of regulated cell death. It is driven by lipid peroxidation caused by the Fe2+-mediated Fenton reaction, and is therefore morphologically and biochemically distinct from previously described cell death processes such as apoptosis, necrosis, and pyroptosis [1, 2]. Both animal models and studies in human specimens support a causative role for dysregulated ferroptosis in a variety of pathological conditions, including cancer [3,4,5]. Accumulating evidence also suggests that sensitivity to ferroptosis determines the responses of cancer cells to conventional cytotoxic, targeted or immunological therapies [6,7,8]. Therefore, single or combined strategies that induce ferroptosis hold great promise as anticancer treatment [6]. Thanks to intensive research over last decade, a number of potential proferroptotic targets have been identified [3]. It is now clear that lipid metabolism, reactive oxygen species (ROS) homeostasis, and iron metabolism lie at the center of the ferroptotic machinery [6, 9]. At the molecular level, the activation of signaling pathways, such as the p53-solute carrier family 7 member 11 (SLC7A11/xCT) and AMPK- acyl-CoA synthase 4 (ACSL4) axes, plays an essential role [10,11,12]. It should be noted that signaling pathways can be spatially organized at microdomains in living cells, which is a highly effective way of controlling cellular behavior and modulating cell fate [13]. However, the spatially aspects of signaling cascades involved in the regulation of ferroptosis are, as yet, undetermined, hindering our understanding of ferroptosis and limiting the potential application of ferroptosis-targeting therapies in the clinic.
铁死亡是最近定义的一种受调节的细胞死亡形式。它是由 Fe 2+介导的 Fenton 反应引起的脂质过氧化驱动的,因此在形态和生物化学上与先前描述的细胞死亡过程(如细胞凋亡、坏死和焦亡)不同 [ 1 , 2 ]。动物模型和人类标本研究都支持铁死亡失调在多种病理状况(包括癌症)中的致病作用[3,4,5 ] 。越来越多的证据还表明,对铁死亡的敏感性决定了癌细胞对传统细胞毒性、靶向或免疫疗法的反应[ 6,7,8 ]。因此,诱导铁死亡的单一或组合策略作为抗癌治疗具有广阔的前景[ 6 ]。由于过去十年的深入研究,已经确定了许多潜在的促凋亡靶标[ 3 ]。现在已经清楚,脂质代谢、活性氧 (ROS) 稳态和铁代谢位于铁死亡机制的中心 [ 6 , 9 ]。在分子水平上,信号通路的激活,例如 p53 溶质载体家族 7 成员 11 (SLC7A11/xCT) 和 AMPK-酰基辅酶 A 合酶 4 (ACSL4) 轴,起着至关重要的作用 [ 10 , 11 , 12] ]。值得注意的是,信号通路可以在活细胞的微域上进行空间组织,这是控制细胞行为和调节细胞命运的高效方法[ 13 ]。 然而,参与铁死亡调节的信号级联的空间方面尚未确定,这阻碍了我们对铁死亡的理解,并限制了铁死亡靶向疗法在临床中的潜在应用。
Mitochondria are a major source of endogenous ROS and host a variety of other metabolic processes, such as ATP production, iron metabolism, and glutaminolysis [14, 15]. It is therefore not surprising that mitochondrial dysfunction has been suggested and recently discovered to be actively involved in ferroptosis [16, 17]. Mitochondria-associated endoplasmic reticulum (ER) membranes (MAMs) are nanodomains of contact sites between mitochondria and the ER that are known to participate in fundamental biological processes, such as lipid and calcium homeostasis, and mitochondrial dynamics [18,19,20]. Therefore, MAMs are not only housekeepers but also signaling hubs. Interestingly, Booth and colleagues recently demonstrate the existence of an H2O2 nanodomain at the ER–mitochondrial interface, suggesting that MAMs may be an essential cellular niche for the transmission of oxidative signals and dealing with oxidative stress [21]. This comes as no surprise because the ER, like mitochondria, is also a primary ROS-producing organelle, and the ER membrane has recently been reported to be a key site of lipid peroxidation [22]. Thus, MAM-confined signaling pathways may be involved in the regulation of ferroptosis.
线粒体是内源性 ROS 的主要来源,并主持各种其他代谢过程,例如 ATP 产生、铁代谢和谷氨酰胺分解 [ 14 , 15 ]。因此,毫不奇怪,线粒体功能障碍已被提出并且最近发现它积极参与铁死亡[ 16 , 17 ]。线粒体相关内质网 (ER) 膜 (MAM) 是线粒体和 ER 之间接触位点的纳米结构域,已知参与基本生物过程,例如脂质和钙稳态以及线粒体动力学 [18,19,20 ] 。因此,MAM不仅是管家,也是信号枢纽。有趣的是,Booth 及其同事最近证明了 ER-线粒体界面上存在 H 2 O 2纳米结构域,这表明 MAM 可能是传递氧化信号和处理氧化应激的重要细胞生态位 [ 21 ]。这并不奇怪,因为内质网和线粒体一样,也是产生 ROS 的主要细胞器,并且最近报道内质网膜是脂质过氧化的关键位点 [ 22 ]。因此,MAM 限制的信号通路可能参与铁死亡的调节。
Protein kinase A (PKA) is a ubiquitously expressed and highly conserved serine/threonine kinase involved in multiple physiological processes [23, 24]. It consists of two regulatory subunits and two catalytic subunits. Cyclic adenosine monophosphate (cAMP) binds to the regulatory subunit and unleashes the catalytic activity of PKA [25]. Notably, PKA activation exhibits a highly spatiotemporal feature. A-kinase anchor proteins (AKAPs) play an essential role in this scenario by tethering PKA and cAMP regulators at the signaling ‘microdomains’ [26]. AKAP1 is the first AKAP identified that is generally considered to be localized at the cytosolic face of the outer mitochondrial membrane (OMM), where it scaffolds PKA to limit the spatial range of PKA phosphorylation of its targets [24, 27, 28]. Recently, AKAP1/PKA has been detected at MAMs, where it regulates lipid metabolism, mitochondria dynamics and the onset of apoptosis [29,30,31,32]. Whether localized AKAP1/PKA signaling components also contribute to oxidative signal transduction and ferroptotic regulation remains unknown.
蛋白激酶A (PKA) 是一种广泛表达且高度保守的丝氨酸/苏氨酸激酶,参与多种生理过程[ 23 , 24 ]。它由两个调节亚基和两个催化亚基组成。环磷酸腺苷 (cAMP) 与调节亚基结合并释放 PKA 的催化活性 [ 25 ]。值得注意的是,PKA 激活表现出高度时空特征。 A-激酶锚定蛋白 (AKAP) 通过将 PKA 和 cAMP 调节因子束缚在信号“微域”上,在这种情况下发挥着重要作用 [ 26 ]。 AKAP1 是第一个被识别的 AKAP,通常被认为位于线粒体外膜 (OMM) 的胞质面,在那里它支撑 PKA 以限制其靶标的 PKA 磷酸化的空间范围 [24,27,28 ] 。最近,AKAP1/PKA 在 MAM 中被检测到,它调节脂质代谢、线粒体动力学和细胞凋亡的开始 [29,30,31,32 ] 。局部 AKAP1/PKA 信号传导成分是否也有助于氧化信号转导和铁死亡调节仍不清楚。
The 75-kDa glucose-regulated protein (GRP75/Mortalin/HSPA9) is a unique HSP70 family member localized primarily in the mitochondrial matrix. GRP75 was initially reported to regulate mitochondrial protein homeostasis by modulating mitochondrial protein import, folding and degradation [33]. Subsequent studies have revealed that GRP75 can be activated in response to oxidative stress, allowing cells to survive under inhospitable oxidative conditions [34, 35]. Inhibition of GRP75 expression leads to mitochondrial protein condensation and mitochondrial dysfunction, characterized by decreased ATP production and increased ROS generation [35, 36]. To complicate matters, GRP75 has also been detected in other cellular compartments such as MAMs and cytosol [37, 38]. As a classical mitochondrial chaperone and key MAM tethering protein, GRP75’s expression level is elevated in many cancers, and increased GRP75 expression is associated with poor prognosis and drug resistance in colon cancer and many other cancer type [33, 39, 40]. It is likely that GRP75 acts as an oncogene by influencing different cellular processes in localization- and context-dependent manners. However, how its subcellular localization is regulated remains largely unexplored, as is its roles in ferroptosis.
75 kDa 葡萄糖调节蛋白 (GRP75/Mortalin/HSPA9) 是一种独特的 HSP70 家族成员,主要位于线粒体基质中。 GRP75 最初被报道通过调节线粒体蛋白输入、折叠和降解来调节线粒体蛋白稳态[ 33 ]。随后的研究表明,GRP75 可以响应氧化应激而被激活,从而使细胞能够在恶劣的氧化条件下生存[ 34 , 35 ]。 GRP75 表达的抑制会导致线粒体蛋白浓缩和线粒体功能障碍,其特征是 ATP 生成减少和 ROS 生成增加 [ 35 , 36 ]。更复杂的是,GRP75 也在其他细胞区室中被检测到,例如 MAM 和细胞质 [ 37 , 38 ]。作为经典的线粒体伴侣和关键的 MAM 束缚蛋白,GRP75 的表达水平在许多癌症中升高,并且GRP75 表达增加与结肠癌和许多其他癌症类型的不良预后和耐药性相关 [ 33,39,40 ]。 GRP75 很可能通过以定位和上下文依赖的方式影响不同的细胞过程而充当癌基因。然而,它的亚细胞定位是如何调节的,以及它在铁死亡中的作用,在很大程度上仍未被探索。
Here, we found that GRP75 is redistributed outside of mitochondria as a result of its increased phosphorylation, which is specifically catalyzed by AKAP1-archored PKA activation at MAMs under ferroptotic conditions. As a result, GRP75 protects cancer cells from ferroptosis by directly binding with Kelch-like ECH-associated protein 1 (Keap1), at MAMs and in the cytosol, leading to reduced Keap1-mediated transcription factor nuclear factor erythroid 2-related factor 2 (Nrf2) degradation and increased expression of a panel of proferroptotic genes downstream of Nrf2. These data expose the spatiotemporal complexity of ferroptosis signaling, and reveal the PKA/GRP75 axis to be a promising treatment target to improve cancer cells’ responses to ferroptosis.
在这里,我们发现 GRP75 由于其磷酸化增加而在线粒体外重新分布,而磷酸化是在铁死亡条件下由 MAM 上 AKAP1 锚定的 PKA 激活特异性催化的。因此,GRP75 通过在 MAM 和细胞质中直接与 Kelch 样 ECH 相关蛋白 1 (Keap1) 结合,从而导致 Keap1 介导的转录因子核因子红细胞 2 相关因子 2 减少,从而保护癌细胞免于铁死亡。 Nrf2) 降解并增加 Nrf2 下游一组促凋亡基因的表达。这些数据揭示了铁死亡信号传导的时空复杂性,并揭示 PKA/GRP75 轴是改善癌细胞对铁死亡反应的有希望的治疗靶点。
Results 结果
GRP75 determines the ferroptotic sensitivity of colon cancers
GRP75 决定结肠癌的铁死亡敏感性
HSP70 family proteins play essential roles in human cancers; however, the underlying mechanisms are not fully understood. By analyzing Clinical Proteomic Tumor Analysis Consortium (CPTAC) data sets, we found that the four most popular heat shock protein 70 (HSP70) genes (GRP75, GRP78, GRP94 and GRP170) were frequently overexpressed at protein levels in multiple types of human cancer (Fig. S1A). Notably, GRP75 levels were more dramatically upregulated in colon cancer than other family members (Fig. 1A and Fig. S1A). Increased GRP75 protein expression in colon cancers was confirmed by western blot analysis of 40 paired colon cancer tissues (Fig. 1B), and increased GRP75 protein expression was associated with advanced stages of the disease (Fig. 1C). The GRP75 gene was also upregulated at the mRNA level in colon cancers, as shown by analyzing The Cancer Genome Atlas (TCGA) dataset (Fig. 1D), and the prognostic value of increased GRP75 mRNA levels was evident (Fig. 1E). We then sought to investigate the key mechanisms of GRP75’s action in colon cancer.
HSP70 家族蛋白在人类癌症中发挥着重要作用;然而,其根本机制尚未完全了解。通过分析临床蛋白质组学肿瘤分析联盟 (CPTAC) 数据集,我们发现四种最流行的热休克蛋白 70 (HSP70) 基因(GRP75、GRP78、GRP94 和 GRP170)在多种人类癌症的蛋白质水平上经常过度表达。图S1A )。值得注意的是,GRP75 水平在结肠癌中比其他家族成员更显着上调(图1A和图S1A )。通过对40对结肠癌组织进行蛋白质印迹分析证实了结肠癌中GRP75蛋白表达的增加(图1B ),并且GRP75蛋白表达的增加与疾病的晚期阶段相关(图1C )。通过分析癌症基因组图谱(TCGA)数据集显示,GRP75基因在结肠癌中的mRNA水平也上调(图1D ),并且GRP75 mRNA水平增加的预后价值是明显的(图1E )。然后我们试图研究 GRP75 在结肠癌中作用的关键机制。
图 1:GRP75 决定结肠癌的铁死亡敏感性。

A Expression of GRP75 protein in colon cancer (T) and the corresponding normal controls (N) in Clinical Proteomic Tumor Analysis Consortium (CPTAC) datasets. B Representative western blot of GRP75 (left) and the quantification of GRP75 bands (right) in 40 pairs of in-house human colon cancer samples (T) and paired adjacent normal controls (N). C The expression of GRP75 protein in colon cancer tissues with different stages. D Expression of GRP75 mRNA in colon cancer (T) and the corresponding normal controls (N) in UALCAN database. E Kaplan-Meier overall survival curves in patients with high and low GRP75 mRNA expression in PrognoScan database. F Go enrichment analysis of differentially expressed genes between the control (si-Ctl) and GRP75 knockdown (KD) (si-GRP75) HCT116 cells. G The relationship between GRP75 expression and drug sensitivity was determined by analysis of the Cancer Treatment Response Portal V2 database. H The efficiencies of CRISPR/Cas9-mediated GRP75 knockout (KO) and re-expression of GRP75 were confirmed by western blot in HCT116 and LoVo stable lines (left). IC50 values of sulfasalazine (SAS) were determined by CCK8 assay. I Cell viability of HCT116 and LoVo cells were determined after treated cells with SAS (2 mM, 24 h) combined with pretreatments of Liproxstatin-1 (Lip-1, 0.3 μM), Z-VAD-FMK (Z-VAD, 10 μM) or Necrosulfonamide (Nec-1, 0.5 μM). Data represents the mean ± SEM of three independent experiments. Values in the control groups were normalized to 1. N.S. not significant; **P < 0.01; ##P < 0.05, compared with the treated controls. (B, C, H, I). See also Figure S1 and S2.
A临床蛋白质组肿瘤分析联盟 (CPTAC) 数据集中 GRP75 蛋白在结肠癌 (T) 和相应正常对照 (N) 中的表达。 B 40 对内部人类结肠癌样本 (T) 和配对的相邻正常对照 (N) 中 GRP75 的代表性蛋白质印迹(左)和 GRP75 带的定量(右)。 C不同分期结肠癌组织中GRP75蛋白的表达情况。 D UALCAN 数据库中结肠癌(T)和相应正常对照(N)中 GRP75 mRNA 的表达。 PrognoScan 数据库中 GRP75 mRNA 高表达和低表达患者的E Kaplan-Meier 总生存曲线。 F Go 对对照 (si-Ctl) 和 GRP75 敲低 (KD) (si-GRP75) HCT116 细胞之间差异表达基因进行富集分析。 G通过分析癌症治疗反应门户 V2 数据库确定 GRP75 表达与药物敏感性之间的关系。 H通过 HCT116 和 LoVo 稳定系(左)中的蛋白质印迹证实了 CRISPR/Cas9 介导的 GRP75 敲除 (KO) 和 GRP75 重新表达的效率。通过CCK8测定测定柳氮磺吡啶(SAS)的IC 50值。 I用 SAS (2 mM, 24 h) 处理细胞并结合 Liproxstatin-1 (Lip-1, 0.3 μM)、Z-VAD-FMK (Z-VAD, 10 μM) 预处理后测定 HCT116 和 LoVo 细胞的细胞活力) 或 Necrosulfonamide (Nec-1, 0.5 μM)。数据代表三个独立实验的平均值±SEM。对照组的值标准化为 1。NS 不显着; ** P < 0.01; ## 与处理对照相比, P < 0.05。 ( B 、 C 、 H 、 I )。 另请参见图S1和S2 。
To this end, we divided the colon cancer tissues in the TCGA database into GRP75-low and GRP75-high groups, using the median value of GRP75 expression as a threshold. Interestingly, in addition to significant enrichment in general cancer-related biological processes (such as cell proliferation and negative regulation of apoptosis), as well as known activities in the regulation of calcium iron homeostasis, the differentially expressed genes (DEGs) between the two groups were involved in pathways such as lipid metabolism and the oxidative stress response (Fig. S1B). These pathways have been demonstrated to affect ferroptosis. We next compared the gene expression profiles of control and GRP75 knockdown (KD) HCT116 colon cancer cells (Fig. S1C). As shown, GRP75 KD significantly altered gene expression profiles (Fig. S1D, E). The DEGs were similarly enriched in biological processes related to ferroptosis, such as ROS metabolic processes, response to iron ions and lipid metabolism (Fig. 1F). In addition, analysis of data from the Cancer Therapeutics Response Portal (CTRP) revealed that higher levels of GRP75 expression were associated with higher IC50 values of mechanistically diverse ferroptosis inducers, including parthenolide, neratinib, ML162, and lapatinib (Fig. 1G). Furthermore, GRP75 expression levels were inversely correlated with lipid peroxidase levels, a driver force of ferroptosis, in a panel of cancer cell lines (Fig. S1F). Therefore, we hypothesized that GRP75 may have a previously unrecognized biological role in protecting cancer cells from ferroptosis, an emerging tumor suppressive mechanism.
为此,我们以GRP75表达的中值作为阈值,将TCGA数据库中的结肠癌组织分为GRP75低组和GRP75高组。有趣的是,除了一般癌症相关生物过程(例如细胞增殖和细胞凋亡的负调节)的显着富集以及钙铁稳态调节的已知活性之外,两组之间的差异表达基因(DEG)参与脂质代谢和氧化应激反应等途径(图S1B )。这些途径已被证明会影响铁死亡。接下来,我们比较了对照和 GRP75 敲低 (KD) HCT116 结肠癌细胞的基因表达谱(图S1C )。如图所示,GRP75 KD 显着改变了基因表达谱(图S1D、E )。 DEGs同样富含与铁死亡相关的生物过程,例如ROS代谢过程、对铁离子的反应和脂质代谢(图1F )。此外,对癌症治疗反应门户(CTRP)数据的分析表明,较高水平的GRP75表达与机械上不同的铁死亡诱导剂(包括小白菊内酯、来拉替尼、ML162和拉帕替尼)较高的IC 50值相关(图1G )。此外,在一组癌细胞系中,GRP75表达水平与脂质过氧化物酶水平呈负相关,脂质过氧化物酶水平是铁死亡的驱动力(图S1F )。因此,我们假设 GRP75 在保护癌细胞免受铁死亡(一种新兴的肿瘤抑制机制)方面可能具有以前未被认识的生物学作用。
To test this possibility, we depleted GRP75 expression by CRISPR/Cas9-mediated knockout (KO) in two commonly used colon cancer cell lines, HCT116 and LoVo, and compared their sensitivities to ferroptosis (Fig. 1H). As shown, GRP75 KO cells exhibited increased sensitivity to ferroptosis induced by mechanistically different reagents such as SAS (sulfasalazine), a USA Food and Drug Administration (FDA)-approved drug for the treatment of inflammatory bowel disease, the xCT inhibitor erastin, and the glutathione peroxidase 4 (GPX4) inhibitors RSL-3 and ML162. Restoration of GRP75 expression abrogated the effect of GRP75 KO (Fig. 1H and S2A). Cell death induced by the above treatments was completely abolished by the ferroptosis inhibitor Lip1 (Fig. 1H, I and Fig. S2A, B), whereas neither treatment with the apoptosis inhibitor Z-VAD nor the necrosis inhibitor Nec-1 was able to do so (Fig. 1I). The anti-ferroptotic effects of endogenous GRP75 were also confirmed by treating cells with two independent small interfering (si)RNA oligos specifically targeting GRP75 in cancer cell lines with different tissue origins, such as HeLa (cervical cancer), 97H (hepatocellular carcinoma), HT1080 (fibrosarcoma) and HT-29 (colon cancer, Fig. S2C). By contrast, GRP75 overexpression attenuated ferroptosis (Fig. S2D, E). These results show that GRP75 plays a general role in protecting cancer cells from ferroptosis.
为了测试这种可能性,我们通过 CRISPR/Cas9 介导的敲除(KO)在两种常用的结肠癌细胞系 HCT116 和 LoVo 中消除了 GRP75 的表达,并比较了它们对铁死亡的敏感性(图1H )。如图所示,GRP75 KO 细胞对机制不同的试剂(例如 SAS(柳氮磺胺吡啶)、美国食品和药物管理局 (FDA) 批准用于治疗炎症性肠病的药物、xCT 抑制剂erastin 和谷胱甘肽过氧化物酶 4 (GPX4) 抑制剂 RSL-3 和 ML162。 GRP75表达的恢复消除了GRP75 KO的作用(图1H和S2A )。上述处理诱导的细胞死亡被铁死亡抑制剂 Lip1 完全消除(图1H、I和图S2A、B ),而凋亡抑制剂 Z-VAD 和坏死抑制剂 Nec-1 均无法消除上述处理引起的细胞死亡。所以(图1I )。内源性 GRP75 的抗铁死亡作用也通过用两种独立的小干扰 (si)RNA 寡核苷酸处理不同组织来源的癌细胞系(如 HeLa(宫颈癌)、97H(肝细胞癌)、 HT1080(纤维肉瘤)和HT-29(结肠癌,图S2C )。相比之下,GRP75 过表达减弱了铁死亡(图S2D、E )。这些结果表明,GRP75 在保护癌细胞免于铁死亡方面发挥着普遍作用。
GRP75 is sequestered outside the mitochondria under ferroptotic conditions
在铁死亡条件下,GRP75 被隔离在线粒体外
Targeting ferroptosis is a promising strategy to eradicate cancer cells. Given the antiferroptotic activity of GRP75 outlined above, we sought to investigate the underlying mechanisms of GRP75 regulation, as this may provide insights into new strategies through which GRP75’s activity can be targeted. We initially speculated that GRP75 expression might be dynamically altered after the induction of ferroptosis. However, GRP75 protein levels were comparable before and after ferroptosis induction (Fig. S3A). If not regulated at the protein level, how is GRP75 activity modulated during ferroptosis? It is well known that the localization of a protein determines its function. GRP75 is a mitochondrial matrix chaperone. Indeed, under basal conditions, GRP75 was predominantly distributed inside the membranous organelle, but not in the released fraction (out of cellular organelle), as revealed by subcellular fractionation described in the materials and methods. Upon stimulation by different ferroptosis inducers, the amount of GRP75 within cellular organelle was reduced to approximately 50% of the control as indicated in the permeabilized cells, which was accompanied by increased GRP75 in the released fraction. The re-distribution of GRP75 was abolished by the ferroptosis inhibitor Lip1 (Fig. 2A and Fig. S3B). Confocal microscopy analysis showed that GRP75 mainly colocalized with HSP60, a mitochondria lumen protein. Remarkably, under SAS-induced ferroptotic conditions, their colocalization was compromised, although GRP75 was still detected in the perimitochondrial region (Fig. 2B). Cell fractionation confirmed that a significant fraction of GRP75 was redistributed from mitochondria to the cytosol under ferroptotic conditions. Further purification of crude mitochondria showed that GRP75 in pure mitochondria was significantly decreased, while MAM-localized GRP75 was accordingly increased (Fig. 2C). This caught our attention because while GRP75 is known to be involved in diverse biological processes in different subcellular regions, how its localization is regulated remains poorly understood.
靶向铁死亡是根除癌细胞的一种有前途的策略。鉴于上述 GRP75 的抗铁死亡活性,我们试图研究 GRP75 调节的潜在机制,因为这可能为靶向 GRP75 活性的新策略提供见解。我们最初推测 GRP75 表达可能在铁死亡诱导后发生动态改变。然而,GRP75蛋白水平在铁死亡诱导之前和之后是相当的(图S3A )。如果不在蛋白质水平上进行调节,那么在铁死亡过程中 GRP75 活性是如何调节的?众所周知,蛋白质的定位决定其功能。 GRP75 是线粒体基质伴侣。事实上,在基础条件下,GRP75 主要分布在膜细胞器内,但不分布在释放的部分(细胞器外)中,如材料和方法中描述的亚细胞分级分离所揭示的。在不同的铁死亡诱导剂的刺激下,细胞器内的 GRP75 量减少至对照的约 50%,如透化细胞中所示,同时释放的部分中的 GRP75 增加。 GRP75的重新分布被铁死亡抑制剂Lip1消除(图2A和图S3B )。共聚焦显微镜分析表明,GRP75 主要与线粒体腔蛋白 HSP60 共定位。值得注意的是,在 SAS 诱导的铁死亡条件下,尽管在线粒体周围区域仍检测到 GRP75,但它们的共定位受到损害(图2B )。细胞分级分离证实,在铁死亡条件下,很大一部分 GRP75 从线粒体重新分配到细胞质中。 粗线粒体的进一步纯化显示,纯线粒体中的GRP75显着减少,而MAM定位的GRP75相应增加(图2C )。这引起了我们的注意,因为虽然已知 GRP75 参与不同亚细胞区域的多种生物过程,但其定位如何调节仍知之甚少。
图 2:在铁死亡下,PKA 激活是在 MAM 上隔离 GRP75 所必需的。

A The release of GRP75 into the cytosol was detected by the cell permeabilization assay in HCT116 cells treated with DMSO, SAS (2 mM, 24 h) with or without Lip-1(0.3 μM)). B Representative confocal microscopy images of GRP75 (Red) and mitochondrial marker HSP60 (Green) in LoVo cells treated with DMSO and SAS (2 mM, 24 h). Scale bar = 10 μm. C The subcellular localization of GRP75 was determined by cell fractionation assay in HCT116 cells treated with DMSO and SAS (2 mM, 24 h). D The phosphorylation of GRP75 was detected in HCT116 cells treated with DMSO or SAS (2 mM, 24 h) in the presence or absence of the PKA inhibitors, H-89 (10 μM, 24 h) and/or KT5720 (5 μm, 24 h). E Potential binding partners of GRP75 were detected by mass spectrum following IP of GRP75 in HCT116 cells treated with DMSO or SAS (2 mM, 24 h). F The PKA activity was detected in HCT116 cells treated with DMSO or SAS (2 mM, 24 h) in the presence or absence of the PKA inhibitors, H-89 (10 μM, 24 h) and/or KT5720 (5 μm, 24 h). G The release of GRP75 into the cytosol was detected in HCT116 cells treated with DMSO or SAS (2 mM, 24 h) in the presence or absence of the PKA inhibitors, H-89 (10 μM, 24 h) and/or KT5720 (5 μm, 24 h). Data represents the Mean ± SEM of three independent experiments. Values in the control groups were normalized to 1. N.S. not significant; *P < 0.05; **P < 0.01; #P < 0.05; ##P < 0.01, compared with the treated controls (A, C, D, F, G). See also Figure S3 and S4.
A在用 DMSO、SAS(2 mM,24 小时)加或不加 Lip-1(0.3 μM)处理的 HCT116 细胞中,通过细胞透化测定检测 GRP75 释放到细胞质中。 B用 DMSO 和 SAS(2 mM,24 小时)处理的 LoVo 细胞中 GRP75(红色)和线粒体标记物 HSP60(绿色)的代表性共聚焦显微镜图像。比例尺 = 10 μm。 C在用 DMSO 和 SAS(2 mM,24 小时)处理的 HCT116 细胞中通过细胞分级分析测定 GRP75 的亚细胞定位。 D在存在或不存在 PKA 抑制剂 H-89(10 μM,24 小时)和/或 KT5720(5 μm,24 小时)的情况下,用 DMSO 或 SAS(2 mM,24 小时)处理的 HCT116 细胞中检测到 GRP75 磷酸化。 24 小时)。 E在用 DMSO 或 SAS(2 mM,24 小时)处理的 HCT116 细胞中对 GRP75 进行 IP 后,通过质谱检测到 GRP75 的潜在结合伴侣。 F在存在或不存在 PKA 抑制剂 H-89(10 μM,24 小时)和/或 KT5720(5 μm,24 小时)的情况下,在用 DMSO 或 SAS(2 mM,24 小时)处理的 HCT116 细胞中检测到 PKA 活性。 h)。 G在存在或不存在 PKA 抑制剂 H-89(10 μM,24 小时)和/或 KT5720 的情况下,用 DMSO 或 SAS(2 mM,24 小时)处理的 HCT116 细胞中检测到 GRP75 释放到细胞质中( 5微米,24小时)。数据代表三个独立实验的平均值±SEM。对照组的值标准化为 1。NS 不显着; * P <0.05; ** P < 0.01; #P <0.05; ## 与处理对照( A、 C 、 D 、 F、G )相比, P < 0.01。另请参见图S3和S4 。
Ferroptosis-induced GRP75 translocation is caused by PKA-mediated GRP75 phosphorylation
铁死亡诱导的 GRP75 易位是由 PKA 介导的 GRP75 磷酸化引起的
Post-translational modification (PTM) is known to modulate the subcellular localization of proteins. We investigated the PTM of GRP75 by immunoprecipitation (IP) of GRP75 followed by western blotting with antibodies that recognize specific modifications of residues, such as acetylation, methylation, ubiquitination, and phosphorylation. As shown, phosphorylation of GRP75 at serine/threonine residues was barely detectable under basal conditions but was significantly increased by ferroptosis, whereas other modifications of GRP75 were either undetectable (acetylation or methylation) or unaffected by ferroptotic conditions (ubiquitination) (1-2 lanes, Fig. 2D and Fig. S3C). We then tried to identify possible serine/threonine kinases by mass spectrometry in the control and ferroptotic HCT116 cells after IP of GRP75. Interestingly, PKA was one of top GRP75 binding partner that was increased in ferroptotic cells compared to control cells (Fig. 2E). We then speculated that PKA may be a major kinase that contributes to the increased phsophorylation of GRP75 (p-GRP75) under ferroptosis. Supportively, the activity of PKA was found to be significantly increased by ferroptosis as demonstrated by kinase activity assay. Pretreatment with the PKA inhibitors, H-89 or KT5720 abolished ferroptosis-induced PKA activation (Fig. 2F), and also reduced the levels of ferroptosis-induced p-GRP75 by approximately 70% in HCT116 cells (3-4 lanes, Fig. 2D). Genetic KD of PKA-Cα by the specific si-RNA oligos also compromised ferroptosis-induced p-GRP75 (Fig. S4A). p-GRP75 was increased by additional ferroptosis inducers, such as RSL-3, erastin and ML162, and similarly repressed by H-89 or KT5720 (Fig. S4B–D). These data suggest that, although it is not the sole kinase, PKA is a major kinase that is activated and subsequently catalyzes GRP75 phosphorylation under ferroptotic conditions. Further subcellular fractionation revealed that H-89 or KT5720 completely blocked ferroptosis-induced GRP75 protein release and redistribution of GRP75 to the released fraction (Fig. 2G). These data suggest that PKA-mediated GRP75 phosphorylation is a prerequisite for the capture GRP75 outside of mitochondria.
已知翻译后修饰 (PTM) 可调节蛋白质的亚细胞定位。我们通过 GRP75 免疫沉淀 (IP),然后使用识别残基特定修饰(如乙酰化、甲基化、泛素化和磷酸化)的抗体进行蛋白质印迹,研究了 GRP75 的 PTM。如图所示,GRP75 在丝氨酸/苏氨酸残基处的磷酸化在基础条件下几乎检测不到,但因铁死亡而显着增加,而 GRP75 的其他修饰要么无法检测到(乙酰化或甲基化),要么不受铁死亡条件(泛素化)影响(1-2 个泳道) ,图2D和图S3C )。然后,我们尝试通过质谱法在 GRP75 IP 后的对照细胞和铁死亡 HCT116 细胞中鉴定可能的丝氨酸/苏氨酸激酶。有趣的是,PKA是最重要的GRP75结合配偶体之一,与对照细胞相比,PKA在铁死亡细胞中增加(图2E )。然后我们推测 PKA 可能是一种主要激酶,有助于铁死亡下 GRP75 (p-GRP75) 磷酸化的增加。激酶活性测定证明,铁死亡可显着增加 PKA 的活性。用 PKA 抑制剂 H-89 或 KT5720 进行预处理,消除了铁死亡诱导的 PKA 激活(图2F ),并且还使 HCT116 细胞中铁死亡诱导的 p-GRP75 水平降低了约 70%(3-4 泳道,图 2F)。 2D )。特定si-RNA寡核苷酸对PKA-Cα的遗传KD也损害了铁死亡诱导的p-GRP75(图S4A )。 p-GRP75 被其他铁死亡诱导剂(如 RSL-3、erastin 和 ML162)增加,并且类似地被 H-89 或 KT5720 抑制(图S4B-D )。 这些数据表明,虽然 PKA 不是唯一的激酶,但它是一种主要激酶,在铁死亡条件下被激活并随后催化 GRP75 磷酸化。进一步的亚细胞分级分离揭示H-89或KT5720完全阻断铁死亡诱导的GRP75蛋白释放以及GRP75重新分配至释放的级分(图2G )。这些数据表明 PKA 介导的 GRP75 磷酸化是在线粒体外捕获 GRP75 的先决条件。
Ferroptosis spatially activates AKAP1/PKA at MAM
铁死亡在 MAM 空间激活 AKAP1/PKA
We further looked how ferroptosis activates PKA. Interestingly, lipid ROS inhibitor Lip-1 inhibited lipid peroxidation, and also abolished ferroptosis-induced cAMP production, PKA activation and p-GRP75 expression (Fig. 3A–D). In addition, inhibition of cAMP production abolished ferroptosis-regulated PKA activation (Fig. 3E). These data suggest that lipid ROS promotes cAMP-dependent PKA activation to induce GRP75 phosphorylation.
我们进一步研究了铁死亡如何激活 PKA。有趣的是,脂质ROS抑制剂Lip-1抑制脂质过氧化,并且还消除了铁死亡诱导的cAMP产生、PKA激活和p-GRP75表达(图3A-D )。此外,cAMP产生的抑制消除了铁死亡调节的PKA激活(图3E )。这些数据表明脂质 ROS 促进 cAMP 依赖性 PKA 激活,从而诱导 GRP75 磷酸化。
图 3:PKA 在 MAM 处磷酸化 GRP75。

A–C Lipid ROS levels (A), cAMP levels (B) and PKA activity (C) were detected in HCT116 cells treated with DMSO, SAS (2 mM, 24 h) with or without Lip-1(0.3 μM). D The phosphorylation of GRP75 was detected in HCT116 cells treated with DMSO or SAS (2 mM, 24 h) in the presence or absence of Lip-1 (0.3 μM). E PKA activity were detected in HCT116 cells treated with DMSO, SAS (2 mM, 24 h) and/or cAMP inhibitor SQ22536 (25 μM, 100 μM). F Representative SIM microscopy images of lipid oxidation (Green) and Endoplasmic reticulum marker Calnexin (CNX) (Red) in HT1080 cells treated with DMSO and SAS (0.5 mM, 24 h) and/or Lip-1(0.3 μM). Scale bar = 10 μm. G The expression of GRP75 and PKA-Cα in the cytosol or MAMs fraction were determined by western blot assay after cell fractionation of the control and AKAP1 KD HCT116 cells treated with DMSO or SAS (2 mM, 24 h). H PKA-Cα/GRP75 binding in the control or AKAP1 knockdown (KD) HCT116 cells treated with DMSO or SAS (2 mM, 24 h) was detected by immunoprecipitation (IP) assay. I The phosphorylation level of GRP75 was determined by immunoprecipitation (IP) of GRP75 followed by western blot assay of anti-phosph-serine/threonine (p-S/T) antibody in the control and AKAP1 KD HCT116 cells treated with DMSO or SAS (2 mM, 24 h). J The release of GRP75 into cytosol was determined by the cell permeabilization assay in the control and AKAP1 KD HCT116 cells treated with DMSO or SAS (2 mM, 24 h). K The diagram illustrates that OMM protein AKAP1 anchored PKA at MAMs, where it phosphorylates GRP75, preventing GRP75 entered in the mitochondria. Data represents the mean ± SEM of three independent experiments. Values in the control groups were normalized to 1. N.S. not significant; **P < 0.01; #P < 0.01; ##P < 0.05, compared with the treated control (A–E, G–J).
A –C在使用或不使用 Lip-1(0.3 μM) 的 DMSO、SAS(2 mM,24 h)处理的 HCT116 细胞中检测脂质 ROS 水平 ( A )、cAMP 水平 ( B ) 和 PKA 活性 ( C )。 D在存在或不存在 Lip-1 (0.3 μM) 的情况下,在用 DMSO 或 SAS(2 mM,24 小时)处理的 HCT116 细胞中检测到 GRP75 的磷酸化。在用 DMSO、SAS(2 mM,24 小时)和/或 cAMP 抑制剂 SQ22536(25 μM,100 μM)处理的 HCT116 细胞中检测到E PKA 活性。 F用 DMSO 和 SAS(0.5 mM,24 小时)和/或 Lip-1(0.3 μM)处理的 HT1080 细胞中脂质氧化(绿色)和内质网标记物钙联蛋白 (CNX)(红色)的代表性 SIM 显微镜图像。比例尺 = 10 μm。 G在用 DMSO 或 SAS(2 mM,24 小时)处理的对照细胞和 AKAP1 KD HCT116 细胞进行细胞分级后,通过蛋白质印迹测定法测定细胞质或 MAM 部分中 GRP75 和 PKA-Cα 的表达。通过免疫沉淀 (IP) 测定检测用 DMSO 或 SAS(2 mM,24 小时)处理的对照或 AKAP1 敲低 (KD) HCT116 细胞中的H PKA-Cα/GRP75 结合。 I GRP75 的磷酸化水平通过 GRP75 的免疫沉淀 (IP) 进行测定,然后对对照中的抗磷酸丝氨酸/苏氨酸 (pS/T) 抗体进行蛋白质印迹测定,以及用 DMSO 或 SAS (2 mM) 处理的 AKAP1 KD HCT116 细胞,24 小时)。 J通过细胞透化测定测定对照细胞和用 DMSO 或 SAS(2 mM,24 小时)处理的 AKAP1 KD HCT116 细胞中 GRP75 释放到细胞质中的情况。 K该图说明 OMM 蛋白 AKAP1 将 PKA 锚定在 MAM 上,磷酸化 GRP75,阻止 GRP75 进入线粒体。 数据代表三个独立实验的平均值±SEM。对照组的值标准化为 1。NS 不显着; ** P < 0.01; #P <0.01; ## 与处理对照( A–E、G–J )相比, P < 0.05。
Both MAM- and non-MAM-PKA were detected in cancer cells under both basal and ferroptotic conditions and no protein translocation was detected (Fig. S4E, F). Since ER-enriched lipid peroxidation was detected after triggering ferroptosis by structured illumination microscopy (SIM) analysis (Fig. 3F), and MAM enrichment of GRP75 was also detected (Fig. 2C), the next question was whether GRP75 is phosphorylated by PKA that is confined to MAMs or not. To clarify this question, we inhibited the expression of AKAP1, an OMM chaperone protein that anchors PKA at MAMs. AKAP1 KD inhibited the expression of AKAP1 by >90% and, as expected, almost completely abrogated the localization of PKA at MAMs (Fig. 3G). Interestingly, AKAP1 KD largely attenuated the GRP75/PKA interaction (Fig. 3H) and also reduced ferroptosis-mediated GRP75 phosphorylation by approximately 70%, similarly to the effect of PKA inhibitor or PKA-Cα KD (Fig. 3I). The enrichment of cytosolic and MAM-localized GRP75 were abolished by AKAP1 KD (Fig. 3J). Taken together, these results show that AKAP1-anchored and MAM-localized PKA plays an essential role in GRP75 phosphorylation, which subsequently leads to the redistribution of GRP75 from inside the mitochondria to MAMs and the cytosol under ferroptotic conditions (Fig. 3K).
在基础和铁死亡条件下,癌细胞中均检测到 MAM-和非 MAM-PKA,并且未检测到蛋白质易位(图S4E、F )。由于通过结构照明显微镜(SIM)分析触发铁死亡后检测到富含ER的脂质过氧化(图3F ),并且还检测到GRP75的MAM富集(图2C ),下一个问题是GRP75是否被PKA磷酸化,从而导致GRP75被磷酸化。是否仅限于 MAM。为了澄清这个问题,我们抑制了 AKAP1 的表达,AKAP1 是一种 OMM 伴侣蛋白,将 PKA 锚定在 MAM 上。 AKAP1 KD 将 AKAP1 的表达抑制>90%,并且如预期的那样,几乎完全消除了 PKA 在 MAM 上的定位(图3G )。有趣的是,AKAP1 KD 很大程度上减弱了 GRP75/PKA 相互作用(图3H ),并且还使铁死亡介导的 GRP75 磷酸化减少了约 70%,与 PKA 抑制剂或 PKA-Cα KD 的作用类似(图3I )。 AKAP1 KD 消除了细胞质和 MAM 定位的 GRP75 的富集(图3J )。总而言之,这些结果表明 AKAP1 锚定和 MAM 定位的 PKA 在 GRP75 磷酸化中发挥重要作用,随后导致 GRP75 在铁死亡条件下从线粒体内部重新分布到 MAM 和细胞质(图3K )。
GRP75 is a substrate of PKA
GRP75 是 PKA 的底物
PKA regulates GRP75 phosphorylation under ferroptosis. We then sought to understand whether GRP75 is a direct substrate of PKA. Consistent with mass spectrum data and the data shown in Fig. 2E, an increased interaction between GRP75 and PKA was detected under ferroptosis (Fig. 4A and Fig. S5A). A direct and ferroptosis-regulated GRP75/PKA interaction was supported by proximity ligation amplification (PLA) assay (Fig. 4B and Fig. S5B, C). Subsequent in vitro IP assay using purified proteins identified key PKA-binding residues on GRP75 in its ATPase domain, fragment (F)2-GRP75, and PKA interacted with GRP75 through its protein kinase domain, F2-PKA (Fig. 4C, D). Protein structure simulation allows visualization of the GRP75/PKA interaction. Interestingly, the model also highlights the importance of the ATPase domain of GRP75 and the protein kinase domain of PKA in the GRP75/PKA interaction (Fig. 4E).
PKA 在铁死亡下调节 GRP75 磷酸化。然后我们试图了解 GRP75 是否是 PKA 的直接底物。与质谱数据和图2E中所示的数据一致,在铁死亡下检测到GRP75和PKA之间的相互作用增加(图4A和图S5A )。邻近连接扩增(PLA)测定支持直接且铁死亡调节的GRP75/PKA相互作用(图4B和图S5B、C )。随后使用纯化蛋白的体外 IP 测定鉴定了 GRP75 ATPase 结构域中的关键 PKA 结合残基,片段 (F)2-GRP75,并且 PKA 通过其蛋白激酶结构域 F2-PKA 与 GRP75 相互作用(图4C,D ) 。蛋白质结构模拟可实现 GRP75/PKA 相互作用的可视化。有趣的是,该模型还强调了GRP75的ATP酶结构域和PKA的蛋白激酶结构域在GRP75/PKA相互作用中的重要性(图4E )。
图4:GRP75是PKA的底物。

A, B GRP75 and PKA-Cα binding was detected by IP assay (A) and PLA (B) assays in HCT116 cells treated with DMSO or SAS (2 mM, 24 h). Scale bar = 10 μm. C, D The bindings between PKA-Cα and Full length (FL) GRP75 or its truncated mutants: fragment (F)1, F2 or F3 (C), and the bindings between GRP75 and FL PKA-Cα and its truncated mutants: F1, F2 or F3 (C) were determined by in vitro IP assay using purified proteins. The domain organizations of GRP75 and PKA-Cα were shown left. E The molecular model of PKA-Cα and GRP75 interaction simulated by HDOCK server. F The GRP75 protein sequence contains the consensus PKA phosphorylation motifs. G In vitro kinase assay was performed to examine the ability of PKA in phosphorylating wild type (WT) GRP75 or S148A using purified proteins in the presence of ATP. H The release of FLAG-tagged GRP75 into the cytosol was detected by the cell permeabilization assay in FLAG-GRP75(WT) or FLAG-GRP75(S148A) overexpression HCT116 cells treated with DMSO or SAS (2 mM, 24 h) I IC50 values of SAS was determined in HCT116 cells with different genetic background as indicated in the figure. Data represents the mean ± SEM of three independent experiments. Values in the control groups were normalized to 1. N.S. not significant; **P < 0.01 (A, B, H, I). See also Figure S5.
A 、 B在用 DMSO 或 SAS(2 mM,24 小时)处理的 HCT116 细胞中通过 IP 测定 ( A ) 和 PLA ( B ) 测定检测 GRP75 和 PKA-Cα 结合。比例尺 = 10 μm。 C 、 D PKA-Cα 与全长 (FL) GRP75 或其截短突变体之间的结合:片段 (F)1、F2 或 F3 ( C ),以及 GRP75 与 FL 之间的结合 PKA-Cα 及其截短突变体:F1 、F2 或 F3 ( C ) 使用纯化蛋白通过体外 IP 测定进行测定。 GRP75 和 PKA-Cα 的结构域组织如左侧所示。 E HDOCK服务器模拟的PKA-Cα和GRP75相互作用的分子模型。 F GRP75 蛋白序列包含共有的 PKA 磷酸化基序。 G进行体外激酶测定,以检查 PKA 在 ATP 存在下使用纯化蛋白磷酸化野生型 (WT) GRP75 或 S148A 的能力。 H在用 DMSO 或 SAS(2 mM,24 h)处理的 FLAG-GRP75(WT) 或 FLAG-GRP75(S148A) 过表达 HCT116 细胞中通过细胞透化测定检测 FLAG 标记的 GRP75 释放到细胞质中I IC 50如图所示,在具有不同遗传背景的 HCT116 细胞中测定了 SAS 值。数据代表三个独立实验的平均值±SEM。对照组的值标准化为 1。NS 不显着; ** P < 0.01( A 、 B 、 H 、 I )。另请参见图S5 。
We further sought to identify the key residues of GRP75 that are regulated by PKA-mediated phosphorylation. Based on the previously reported PKA phosphorylation motifs R-X-pS and R-X-pT [41], two conserved PKA phosphorylation residues were predicted in GRP75: S148 and T462 (Fig. 4F and Fig. S5D). Ferroptosis-induced phosphorylation of GRP75 in cells was compromised by mutation of S148A, but not by mutation of T462A (Fig. S5E). Consistently, the PKA inhibitor H-89 reduced the phosphorylation levels of WT GRP75 but had no effect on the phosphorylation levels of S148A GRP75 (Fig. S5F). In vitro kinase assay using purified proteins further showed that PKA phosphorylated wild-type (WT) GRP75, but not GRP75 S148A (Fig. 4G). These data suggest that GRP75 is a substrate of PKA and that the conserved S148 residue is the major phosphorylated residue.
我们进一步试图鉴定受 PKA 介导的磷酸化调节的 GRP75 关键残基。基于先前报道的PKA磷酸化基序RX-pS和RX-pT[ 41 ],预测GRP75中存在两个保守的PKA磷酸化残基:S148和T462(图4F和图S5D )。细胞中铁死亡诱导的GRP75磷酸化受到S148A突变的损害,但不受T462A突变的损害(图S5E )。一致地,PKA抑制剂H-89降低WT GRP75的磷酸化水平,但对S148A GRP75的磷酸化水平没有影响(图S5F )。使用纯化蛋白的体外激酶测定进一步显示PKA磷酸化野生型(WT)GRP75,但不磷酸化GRP75S148A(图4G )。这些数据表明GRP75是PKA的底物并且保守的S148残基是主要的磷酸化残基。
We then investigated the effects of p-S148 on the localization and antiferroptotic activity of GRP75. As shown, ferroptosis promoted WT and irrelevant mutation T462A GRP75 released from intracellular organelle (Fig. S5G), whereas it failed to influence the distribution of GRP75 S148A protein (Fig. 4H). In addition, WT GRP75 abrogated GRP75 KO-promoted ferroptosis, while expression of similar levels of GRP75 S148A in GRP75 KO cells did not (Fig. 4I). Taken together, these data suggest that PKA-mediated phosphorylation of GRP75 at S148 keeps GRP75 outside of the mitochondria and then produces a significant antiferroptotic function.
然后我们研究了 p-S148 对 GRP75 的定位和抗铁死亡活性的影响。如图所示,铁死亡促进WT和无关突变T462A GRP75从细胞内细胞器释放(图S5G ),但它未能影响GRP75 S148A蛋白的分布(图4H )。此外,WT GRP75消除了GRP75 KO促进的铁死亡,而GRP75 KO细胞中类似水平的GRP75 S148A的表达则没有(图4I )。总而言之,这些数据表明 PKA 介导的 GRP75 在 S148 处的磷酸化使 GRP75 保持在线粒体之外,然后产生显着的抗铁死亡功能。
GRP75 protects cells from ferroptosis by interacting with Keap1 to stabilize Nrf2 protein
GRP75 通过与 Keap1 相互作用稳定 Nrf2 蛋白来保护细胞免于铁死亡
We then investigated how GRP75 protects cells from ferroptosis outside of mitochondria. To this end, GRP75 binding proteins identified by mass spectrometry shown in Fig. 2E were further analyzed. Interestingly, in addition to PKA, Keap1 was among the top hits of the differential binding partners before/after triggering ferroptosis. This result was interesting because Keap1 is a known master inhibitor of Nrf2, a previously identified inhibitor of ferroptosis both in vivo and in vitro [42, 43]. Subsequent IP assay confirmed GRP75/Keap1 binding (Fig. S6A). GRP75/Keap1 binding was increased in ferroptotic cells at both the cytosol and the MAMs, and was accompanied by decreased Nrf2/Keap1 binding and increased Nrf2 protein expression (Fig. 5A). Consistent with above data, Keap1 was predominately localized outside of membranous organelles (Fig. S6B). Similarly to endogenous GRP75, exogenous WT GRP75 bound more efficiently to Keap1 in SAS-treated cells than in control cell, while the mutants containing S148A bound with Keap1 relatively weaker than WT or T462A GRP75 regardless of SAS. The different binding abilities of S148A, WT, and T462A GRP75 were not caused by differences in protein levels, as similar amount of GRP75 proteins were detected by western blot analysis using an anti-FLAG tag (Fig. S6C).
然后我们研究了 GRP75 如何保护细胞免受线粒体外的铁死亡的影响。为此,进一步分析了图2E所示的通过质谱鉴定的GRP75结合蛋白。有趣的是,除了 PKA 之外,Keap1 也是触发铁死亡之前/之后差异结合伴侣的热门点击之一。这个结果很有趣,因为 Keap1 是 Nrf2 的已知主要抑制剂,Nrf2 是先前在体内和体外鉴定的铁死亡抑制剂 [ 42 , 43 ]。随后的 IP 测定证实了 GRP75/Keap1 结合(图S6A )。 GRP75/Keap1结合在铁死亡细胞的胞浆和MAM中均增加,并伴随着Nrf2/Keap1结合的减少和Nrf2蛋白表达的增加(图5A )。与上述数据一致,Keap1主要位于膜细胞器之外(图S6B )。与内源性 GRP75 类似,在 SAS 处理的细胞中,外源性 WT GRP75 比对照细胞更有效地与 Keap1 结合,而无论 SAS 如何,含有 S148A 的突变体与 Keap1 的结合相对弱于 WT 或 T462A GRP75。 S148A、WT和T462A GRP75的不同结合能力不是由蛋白质水平的差异引起的,因为使用抗FLAG标签通过蛋白质印迹分析检测到相似量的GRP75蛋白质(图S6C )。
图 5:GRP75 通过与 Keap1 相互作用稳定 Nrf2 蛋白,从而保护细胞免于铁死亡。

A The bindings between Keap1, GRP75 and Nrf2 in the fractionated HCT116 cells treated with DMSO or SAS (2 mM, 24 h) were detected by IP assay in HCT116 cells. B The correlation between GRP75 and Nrf2, GRP75 and Keap1 protein levels in human colon cancer cell lines in Cancer Cell Line Encyclopedia (CCLE) database. C The ubiquitinated (Ub)-Nrf2 was detected by IP of Nrf2 and western blot of anti-HA in the control or GRP75 KD HCT116 cells treated with DMSO or SAS (2 mM, 24 h). All cells are transfected with Ub-HA and the experiments were performed in the presence of MG132 (20 μM). The expression levels of Nrf2 and Ub-Nrf2 were quantified in the bar graph bellow. D The expression of Nrf2 was detected after transfection of FLAG- GRP75(WT) or FLAG-GRP75(S148A) expression plasmids in HCT116 cells in the presence of DMSO or SAS (2 mM, 24 h). E The luciferase activity of the Nrf2-ARE luciferase reporter in the control or GRP75 KD HCT116 cells treated with DMSO or SAS (2 mM, 24 h). F The expression levels of Nrf2 target genes were detected by qRT-PCR in GRP75 KO and/or Nrf2 KD HCT116 cells. G IC50 values of SAS were determined in GRP75 KO and/or Nrf2 KD HCT116 cells. Data represents the mean ± SEM of three independent experiments. Values in the control groups were normalized to 1. N.S. not significant; *P < 0.05; **P < 0.01; #P < 0.05; ##P < 0.01, compared with the treated control (A, C-E, G). See also Figure S6.
A在 HCT116 细胞中通过 IP 测定检测经 DMSO 或 SAS(2 mM,24 小时)处理的分级分离的 HCT116 细胞中 Keap1、GRP75 和 Nrf2 之间的结合。 B癌细胞系百科全书(CCLE)数据库中人结肠癌细胞系中 GRP75 与 Nrf2、GRP75 与 Keap1 蛋白水平的相关性。 C在对照或用 DMSO 或 SAS(2 mM,24 小时)处理的 GRP75 KD HCT116 细胞中,通过 Nrf2 的 IP 和抗 HA 的蛋白质印迹检测泛素化 (Ub)-Nrf2。所有细胞均用 Ub-HA 转染,实验在 MG132 (20 μM) 存在的情况下进行。 Nrf2 和 Ub-Nrf2 的表达水平在下面的条形图中进行了量化。 D在 DMSO 或 SAS 存在下(2 mM,24 h)将 FLAG-GRP75(WT)或 FLAG-GRP75(S148A)表达质粒转染 HCT116 细胞后检测 Nrf2 的表达。 E用 DMSO 或 SAS(2 mM,24 小时)处理的对照或 GRP75 KD HCT116 细胞中 Nrf2-ARE 荧光素酶报告基因的荧光素酶活性。 F通过 qRT-PCR 检测 GRP75 KO 和/或 Nrf2 KD HCT116 细胞中 Nrf2 靶基因的表达水平。在GRP75 KO和/或Nrf2 KD HCT116细胞中测定SAS的G IC 50值。数据代表三个独立实验的平均值±SEM。对照组的值标准化为 1。NS 不显着; * P <0.05; ** P < 0.01; #P <0.05; ## P < 0.01,与处理对照( A 、 CE 、 G )相比。另请参见图S6 。
Keap1 is known to inhibit Nrf2 by inducing its proteasome-mediated degradation by interacting with Nrf2. Interestingly, analysis of Cancer Cell Line Encyclopedia (CCLE) database revealed a positive association between GRP75 and Nrf2 proteins, and no such correlation was detected between GRP75 and Keap1 in human colon cancer cell lines (Fig. 5B). GRP75 KD led to decreased expression of Nrf2 protein but not of its mRNA, which was coordinated with increased Nrf2 ubiquitination under both basal and ferroptotic conditions (Fig. 5C and Fig. S6D, E). In contrast, WT but not S148A GRP75 overexpression increased Nrf2 expression (Fig. 5D). GRP75 KD-induced Nrf2 repression was accompanied by decreased transcriptional activity of Nrf2, as demonstrated by antioxidant responsive element (ARE) luciferase reporter assay (Fig. 5E). A panel of ferroptosis-related target genes, involved in iron, glutathione, and lipid metabolism, were simultaneously inhibited by GRP75 KO in a Nrf2-dependent manner (Fig. 5F). Similar changes regarding SAS-induced ferroptosis and lipid ROS levels were also detected following GRP75 KO with or without Nrf2 KD (Fig. 5G and Fig. S6F). The impacts of GRP75 KO on the expression of key anti-ferroptotic targets of Nrf2 targets, GPX4, FTH1 and FSP1,were further validated at protein levels (Fig. S6G). These results indicate that upon ferroptotic stimulation, GRP75 translocates to the MAM and cytosol, where it binds to Keap1 and protects Nrf2 from Keap1-mediated protein degradation, leading to attenuated ferroptosis.
已知 Keap1 通过与 Nrf2 相互作用诱导其蛋白酶体介导的降解来抑制 Nrf2。有趣的是,癌细胞系百科全书(CCLE)数据库的分析揭示了GRP75和Nrf2蛋白之间的正相关性,并且在人结肠癌细胞系中GRP75和Keap1之间没有检测到这种相关性(图5B )。 GRP75 KD导致Nrf2蛋白的表达降低,但不降低其mRNA的表达,这与基础条件和铁死亡条件下Nrf2泛素化的增加相协调(图5C和图S6D,E )。相反,WT而非S148A GRP75过表达增加了Nrf2表达(图5D )。 GRP75 KD诱导的Nrf2抑制伴随着Nrf2转录活性的降低,如抗氧化反应元件(ARE)荧光素酶报告基因测定所证明的(图5E )。一组涉及铁、谷胱甘肽和脂质代谢的铁死亡相关靶基因同时被GRP75 KO以Nrf2依赖性方式抑制(图5F )。在有或没有Nrf2 KD的情况下GRP75 KO后,也检测到关于SAS诱导的铁死亡和脂质ROS水平的类似变化(图5G和图S6F )。 GRP75 KO 对 Nrf2 靶标 GPX4、FTH1 和 FSP1 的关键抗铁死亡靶标表达的影响在蛋白质水平上得到了进一步验证(图S6G )。这些结果表明,在铁死亡刺激下,GRP75 易位到 MAM 和细胞质,在那里它与 Keap1 结合并保护 Nrf2 免受 Keap1 介导的蛋白质降解,从而导致铁死亡减弱。
GRP75 competes with Nrf2 to binding Keap1 thought its high affinity DLG-binding motif, ETGE
GRP75 与 Nrf2 竞争结合 Keap1,认为其高亲和力 DLG 结合基序 ETGE
We further investigated the molecular basis of the GRP75-regulated Keap1/Nrf2 axis in ferroptosis. First, direct interaction between GRP75/Keap1 was observed by PLA assays in cells (Fig. 6A) and in vitro IP assays using recombinant proteins, while there was no direct interaction between GRP75 and Nrf2 (Fig. 6B). GRP75 dose-dependently disrupted Keap1/Nrf2 binding (Fig. 6C), suggesting that GRP75 may compete with Nrf2 for Keap1 binding. Nrf2 is known to bind to the RDG motif in Keap1 through two distinct motifs with different affinities: a high-affinity ETGE motif and a low-affinity DLG motif [44]. Interestingly, we identified two DLG motifs and one ETGE motif in GRP75, all of which are highly conserved across species (Fig. 6D). The alpha-fold-predicted GRP75 structure indicated that the ETGE motif is distributed on the protein surface, whereas the DLG motif is embedded inside the protein structure (Fig. 6E). Indeed, mutation in the ETGE motif in GRP75 completely abolished the ability of GRP75 to bind to Keap1, whereas mutation in the DLG motif had no obvious effect (Fig. 6F). Collectively, our results demonstrate that pS148-GRP75 is enriched in the MAMs and cytosol, where it competes with Nrf2 for Keap1 binding through its high-affinity ETGE motif, and thus protects Nrf2 from Keap1-mediated protein degradation.
我们进一步研究了铁死亡中 GRP75 调节的 Keap1/Nrf2 轴的分子基础。首先,通过细胞中的PLA测定(图6A )和使用重组蛋白的体外IP测定观察到GRP75/Keap1之间的直接相互作用,而GRP75和Nrf2之间没有直接相互作用(图6B )。 GRP75剂量依赖性地破坏Keap1/Nrf2结合(图6C ),表明GRP75可能与Nrf2竞争Keap1结合。已知 Nrf2 通过具有不同亲和力的两个不同基序与 Keap1 中的 RDG 基序结合:高亲和力 ETGE 基序和低亲和力 DLG 基序 [ 44 ]。有趣的是,我们在GRP75中鉴定了两个DLG基序和一个ETGE基序,所有这些基序在物种间高度保守(图6D )。 α折叠预测的GRP75结构表明ETGE基序分布在蛋白质表面,而DLG基序嵌入蛋白质结构内部(图6E )。事实上,GRP75中ETGE基序的突变完全消除了GRP75结合Keap1的能力,而DLG基序的突变没有明显的影响(图6F )。总的来说,我们的结果表明 pS148-GRP75 在 MAM 和细胞质中富集,通过其高亲和力 ETGE 基序与 Nrf2 竞争 Keap1 结合,从而保护 Nrf2 免受 Keap1 介导的蛋白质降解。
图 6:GRP75 与 Nrf2 竞争与 Keap1 的结合(认为是高亲和力 ETGE 基序)。

A The interaction between GRP75 and Keap1 was determined by PLA assay in HCT 116 treated with DMSO or SAS (2 mM, 24 h). Scale bar = 10 μm. B The binding between GRP75, Keap1 and Nrf2 were determined by in vitro IP assay using purified proteins. C The interactions between Keap1 and Nrf2 or GRP75 were evaluated with increasing dose of Grp75 protein by in vitro IP assays. The bands of Keap1, GRP75, and Nrf2 were quantified by Image J and the quantification data were shown in line graph. D The conservation of ETGE and DLG motif in GRP75 protein sequence in multiple species. E The protein structure of GRP75 was predicted by alpha Fold. F The binding between the Keap1 and WT or mutant M1, M2 or M3 GRP75 was measured by IP assay. Data represents the mean ± SEM of three independent experiments. Values in the control groups were normalized to 1. **P < 0.01 (A).
A通过在用 DMSO 或 SAS(2 mM,24 小时)处理的 HCT 116 中进行 PLA 测定来确定 GRP75 和 Keap1 之间的相互作用。比例尺 = 10 μm。 B使用纯化蛋白通过体外 IP 测定法测定 GRP75、Keap1 和 Nrf2 之间的结合。 C通过体外 IP 测定,随着 Grp75 蛋白剂量的增加,评估 Keap1 与 Nrf2 或 GRP75 之间的相互作用。 Keap1、GRP75和Nrf2的条带通过Image J进行定量,定量数据以线形图显示。 D多个物种的 GRP75 蛋白序列中 ETGE 和 DLG 基序的保守性。 E通过 alpha Fold 预测 GRP75 的蛋白质结构。 F通过 IP 测定法测量 Keap1 与 WT 或突变体 M1、M2 或 M3 GRP75 之间的结合。数据代表三个独立实验的平均值±SEM。对照组的值标准化为 1。** P < 0.01 ( A )。
Inhibition of PKA/GRP75/Nrf2 sensitizes cancer cell responses to SAS-induced ferroptosis
PKA/GRP75/Nrf2 的抑制使癌细胞对 SAS 诱导的铁死亡反应敏感
Given the essential role of the PKA/GRP75 axis in regulating cellular sensitivity to ferroptosis, we examined whether inhibition of the PKA/GRP75/Nrf2 axis could be utilized as a strategy to treat cancers by inducing ferroptosis. To this end, we generated a HCT116 cell line-derived xenograft (CDX) model with different GRP75 expression levels (Fig. 7A), and the CDX-bearing mice were treated as indicated in Fig. 7B. The results revealed that the growth of GRP75 KO xenografts was relatively slower than that of control xenografts (Fig. 7C–E). Treatment with SAS repressed tumor growth in both control and GRP75 KO xenografts, with no detectable effects on the body weights of mice (Fig. 7F). GRP75 KO significantly increased the sensitivity of xenografts to SAS treatment (Fig. 7C–E). Levels of 4-hydroxynonenal (4-HNE), a lipid peroxidation product and ferroptosis marker, were increased by SAS treatment and further elevated by GRP75 KD (Fig. 7G). In contrast, levels of Ki67, a molecular marker of cell proliferation, were significantly reduced after SAS treatment and further decreased after GRP75 KO (Fig. 7G). Interestingly, re-expression of WT GRP75 abolished the effects of GRP75 KD on tumor growth, and 4-HNE and Ki67 expression, while GRP75-S148A had no detectable effects on them (Fig. 7C–E, G). Western blot analysis of dissected CDX tissue revealed that SAS treatment had little effect on the protein levels of GRP75 and Keap1. However, Nrf2 protein levels increased following SAS treatment and were significantly repressed by GRP75 KO, which was restored by WT GRP75 but not GRP75-S148 A (Fig. 7H). These data suggest that p-S148-GRP75 plays essential roles in protecting cancer cells from ferroptosis.
鉴于 PKA/GRP75 轴在调节细胞对铁死亡的敏感性中的重要作用,我们研究了是否可以利用抑制 PKA/GRP75/Nrf2 轴作为通过诱导铁死亡来治疗癌症的策略。为此,我们生成了具有不同GRP75表达水平的HCT116细胞系来源的异种移植物(CDX)模型(图7A ),并且如图7B所示处理携带CDX的小鼠。结果显示,GRP75 KO异种移植物的生长速度比对照异种移植物相对慢(图7C-E )。用SAS治疗抑制了对照和GRP75 KO异种移植物中的肿瘤生长,对小鼠的体重没有可检测到的影响(图7F )。 GRP75 KO显着增加了异种移植物对SAS治疗的敏感性(图7C-E )。 4-羟基壬烯醛(4-HNE)(一种脂质过氧化产物和铁死亡标志物)的水平通过SAS处理而增加,并通过GRP75 KD进一步升高(图7G )。相反,Ki67(细胞增殖的分子标志物)的水平在SAS处理后显着降低,并且在GRP75 KO后进一步降低(图7G )。有趣的是,WT GRP75的重新表达消除了GRP75 KD对肿瘤生长以及4-HNE和Ki67表达的影响,而GRP75-S148A对它们没有可检测到的影响(图7C-E,G )。对解剖 CDX 组织的蛋白质印迹分析表明,SAS 处理对 GRP75 和 Keap1 的蛋白水平几乎没有影响。然而,SAS处理后Nrf2蛋白水平增加,并被GRP75 KO显着抑制,WT GRP75而非GRP75-S148 A恢复了Nrf2蛋白水平(图7H )。 这些数据表明 p-S148-GRP75 在保护癌细胞免于铁死亡方面发挥着重要作用。
图 7:GRP75 的基因抑制使癌细胞对 CDX 模型中 SAS 诱导的铁死亡的反应敏感。

A GRP75 KO HCT116 cells were generated by CRISPR-Cas9 technique following single clone section. B The schematic diagram of the treatment strategies for the xenograft bearing nude mice. C–E Tumor volumes in the indicated time periods (C), tumor images (D) or tumor weight (E) of HCT116 cell line-derived xenograft (CDX) were presented. F The body weight of nude mice was measured in the indicated time. G, H The expression levels of 4-HNE and Ki67 (G) and GRP75/Keap1/Nrf2 (H) were determined by immunohistochemistry (IHC) and western blot, respectively, in the indicated xenografts. Data represents the mean ± SD (n = 5 for each group). *P < 0.05; **P < 0.01; N.S. not significant; # P < 0.05; ##P < 0.01, compared with the treated controls; $$P < 0.01, compared with the treated GRP75 KO xenograft (C, E–G).
通过单克隆切片后的 CRISPR-Cas9 技术生成GRP75 KO HCT116 细胞。 B裸鼠异种移植治疗策略示意图。 C – E呈现了 HCT116 细胞系来源的异种移植物 (CDX) 在指定时间段 (C) 的肿瘤体积 ( C )、肿瘤图像 ( D ) 或肿瘤重量 ( E )。 F在指定时间内测量裸鼠的体重。 G 、 H分别通过免疫组织化学 (IHC) 和蛋白质印迹测定所示异种移植物中 4-HNE 和 Ki67 ( G ) 和 GRP75/Keap1/Nrf2 ( H ) 的表达水平。数据代表平均值±SD(每组 n = 5)。 * P <0.05; ** P < 0.01; NS 不显着; #P <0.05; ## 与处理对照相比, P < 0.01; $$ P < 0.01,与处理的 GRP75 KO 异种移植物相比 ( C, E – G )。
To explore the potential clinical relevance of these findings, we established a patient-derived xenograft (PDX) model using fresh postoperative colon cancer samples and then treated the tumor-bearing mice with SAS (Fig. 8A). SAS alone has mild effects on tumor growth, as did the GRP75 inhibitor MKT077. However, combination treatment led to robust inhibition of tumor growth (Fig. 8B–D and Fig. S7A–C). Increased levels of 4-HNE were also observed after combination treatment. The ferroptosis inhibitor Lip1 abolished the effects of SAS/MKT077 combination treatment on tumor growth and 4-HNE expression. Levels of the proliferation marker Ki67 were negatively correlated with those of 4-HNE (Fig. 8E and Fig. S7D). The body weights of the tumor-bearing mice did not change as a result of treatment, suggesting that the treatments were well tolerated (Fig. 8F and Fig. S7E). In addition, western blot analysis of dissected PDX samples revealed that the protein expression levels of GRP75 and Keap1 were also not altered by treatment. Interestingly, MKT077 treatment significantly weakened SAS-induced Nrf2 protein accumulation, an effect that was reversed by Lip-1 treatment (Fig. 8G and Fig. S7F).
为了探索这些发现的潜在临床相关性,我们使用新鲜的术后结肠癌样本建立了患者来源的异种移植(PDX)模型,然后用SAS治疗荷瘤小鼠(图8A )。单独的 SAS 对肿瘤生长有轻微影响,GRP75 抑制剂 MKT077 也有同样的效果。然而,联合治疗导致肿瘤生长的强烈抑制(图8B-D和图S7A-C )。联合治疗后还观察到 4-HNE 水平升高。铁死亡抑制剂 Lip1 消除了 SAS/MKT077 联合治疗对肿瘤生长和 4-HNE 表达的影响。增殖标志物Ki67的水平与4-HNE的水平呈负相关(图8E和图S7D )。荷瘤小鼠的体重没有因治疗而改变,表明治疗具有良好的耐受性(图8F和图S7E )。此外,对解剖的 PDX 样本进行蛋白质印迹分析表明,GRP75 和 Keap1 的蛋白表达水平也没有因治疗而改变。有趣的是,MKT077处理显着削弱了SAS诱导的Nrf2蛋白积累,这一效应被Lip-1处理逆转(图8G和图S7F )。
图 8:GRP75 的化学抑制可抑制 PDX 模型中癌细胞对 SAS 诱导的铁死亡的反应。

A The schematic diagram of the treatment strategies for the patient-derived xenograft (PDX) bearing NSG mice. B–D PDX xenograft volumes in the indicated time periods (B), tumor weight (C) or tumor images (D) were presented. E, G The expression levels of 4-HNE and Ki67 (E) and GRP75/Keap1/Nrf2 (G) were determined by IHC and western blot, respectively, in the indicated xenografts. F The body weight of NSG mice were measured in the indicated time. Data represents the mean ± SD (n = 5 for each group). N.S. not significant; *P < 0.05; **P < 0.01 (B, C, E–G). See also Figure S7.
A携带 NSG 小鼠的患者来源异种移植物 (PDX) 治疗策略示意图。 B – D显示指定时间段内的 PDX 异种移植物体积 ( B )、肿瘤重量 ( C ) 或肿瘤图像 ( D )。 E 、 G在指定的异种移植物中,分别通过 IHC 和蛋白质印迹测定 4-HNE 和 Ki67 ( E ) 和 GRP75/Keap1/Nrf2 ( G ) 的表达水平。 F在指定时间内测量 NSG 小鼠的体重。数据代表平均值±SD(每组n = 5)。 NS 不显着; * P <0.05; ** P < 0.01( B 、 C 、 E – G )。另请参见图S7 。
Discussion 讨论
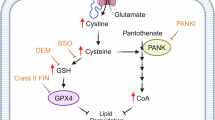
In addition to creating functionally specialized spaces (cellular organelles), intracellular membrane systems provide surfaces for many vital biochemical processes, allowing efficient and rapid responses to dynamically changing local cues. We found that GRP75 is specifically phosphorylated at MAMs by a signaling microdomain of AKAP1-anchored PKA in response to ferroptosis-induced lipid peroxidation. p-GRP75 is unable to enter the mitochondria and, as a result, competes with Nrf2 to bind Keap1 at MAMs and in the cytosol, resulting in increased Nrf2 stabilization and protection against ferroptosis (Fig. 9).
除了创建功能专门的空间(细胞器)之外,细胞内膜系统还为许多重要的生化过程提供表面,从而能够对动态变化的局部线索做出有效和快速的反应。我们发现 GRP75 在 MAM 上被 AKAP1 锚定的 PKA 的信号微结构域特异性磷酸化,以响应铁死亡诱导的脂质过氧化。 p-GRP75 无法进入线粒体,因此与 Nrf2 竞争在 MAM 和细胞质中结合 Keap1,从而增强 Nrf2 的稳定性并防止铁死亡(图9 )。
图 9:MAM 处 AKAP1/PKA/GRP75 局部激活保护细胞免遭铁死亡的模型。

GRP75 is specifically phosphorylated by AKAP1-anchored PKA microdomains on MAMs in response to ferroptosis-induced lipid peroxidation. P-GRP75 cannot enter the mitochondria and therefore competes with Nrf2 to bind Keap1 in MAMs and cytosol, thereby increasing Nrf2’s stability and protection against ferroptosis.
GRP75 被 MAM 上 AKAP1 锚定的 PKA 微结构域特异性磷酸化,以响应铁死亡诱导的脂质过氧化。 P-GRP75 无法进入线粒体,因此与 Nrf2 竞争结合 MAM 和细胞质中的 Keap1,从而增加 Nrf2 的稳定性并防止铁死亡。
Consistent with previous reports, our data show that lipid peroxidation initially occurs mainly on the ER membrane. AKAP1 is anchored to the OMM [28]. It is possible that mitochondria-anchored AKAP1-PKA is activated at the mitochondrial/ER interface (MAMs) due to the spatial proximity of the increased lipid ROS. Interesting, PKA inhibitor and AKAP1 inhibited p-GRP75 to similar extent, suggesting that PKA-mediated GRP75 phosphorylation occurs predominantly at the MAMs in an AKAP1-dependent manner. This spatially restricted phosphorylation may be due to the recently reported phase separation of the regulatory domain in PKA, which limits cAMP diffusion [45]. In addition, GRP75 phosphorylation is undetectable under basal conditions, but GRP75/PKA binding is evident. This may be caused by the low sensitivity of the phosphorylation assay. However, these data may also suggest that substrate and kinase binding is necessary, but not sufficient, for the catalytic reaction to occur. Indeed, non-MAM PKA is detected under both basal and ferroptotic conditions. AKAP1 KD reduces, but isn’t able to completely abolish the binding between PKA and GRP75. We also found that ferroptosis-induced PKA activation is cAMP-dependent. This differs from the recent report that ROS activate PKA in a cAMP-independent manner [46]. This discrepancy suggests that the oxidative response is a highly diverse and tightly regulated process that is spatially and temporally specific, and may also depend on the type of ROS. To complicate matters further, our data suggest that other kinases besides PKA are involved in ferroptosis-induced GRP75 phosphorylation under ferroptotic conditions. These events are unlikely to be important for the translocation of GRP75 from the mitochondria to MAMs and the cytosol. However, future studies are required to understand the differential regulation of GRP75 phosphorylation in ferroptosis.
与之前的报道一致,我们的数据表明脂质过氧化最初主要发生在内质网膜上。 AKAP1 锚定于 OMM [ 28 ]。由于脂质 ROS 增加的空间接近性,线粒体锚定的 AKAP1-PKA 可能在线粒体/内质网界面 (MAM) 处被激活。有趣的是,PKA 抑制剂和 AKAP1 对 p-GRP75 的抑制程度相似,表明 PKA 介导的 GRP75 磷酸化主要以 AKAP1 依赖性方式发生在 MAM 处。这种空间受限的磷酸化可能是由于最近报道的 PKA 中调节域的相分离,这限制了 cAMP 扩散 [ 45 ]。此外,在基础条件下检测不到 GRP75 磷酸化,但 GRP75/PKA 结合是明显的。这可能是由于磷酸化测定的灵敏度低造成的。然而,这些数据也可能表明底物和激酶的结合对于催化反应的发生是必要的,但还不够。事实上,非 MAM PKA 在基础条件和铁死亡条件下均可检测到。 AKAP1 KD 降低,但不能完全消除 PKA 和 GRP75 之间的结合。我们还发现铁死亡诱导的 PKA 激活是 cAMP 依赖性的。这与最近报道的 ROS 以不依赖于 cAMP 的方式激活 PKA 不同 [ 46 ]。这种差异表明氧化反应是一个高度多样化且严格调控的过程,具有空间和时间特异性,并且还可能取决于 ROS 的类型。更复杂的是,我们的数据表明,除 PKA 之外的其他激酶也参与铁死亡条件下铁死亡诱导的 GRP75 磷酸化。 这些事件对于 GRP75 从线粒体到 MAM 和细胞质的易位不太重要。然而,未来的研究需要了解铁死亡中 GRP75 磷酸化的差异调节。
In addition, a known function of GRP75 at MAMs is to tether the ER and mitochondria by interacting with VDAC1 in the OMM and IP3R at the ER membrane [47]. Several regulators, such as TG2 [48] and DJ-1 [49] have been identified to modulate MAM formation by interacting with GRP75. Recently, PDK4-mediated GRP75 phosphorylation has also been shown to promote MAM formation in alcohol-associated liver disease [38]. Our data show that ferroptosis-induced GRP75 phosphorylation by PKA results in GRP75 being locked outside the mitochondria. Residue S148 is located far from the mitochondrial localization signal (1–46 amino acids, aa) in the primary sequence of the GRP75 protein. Their relative spatial positions in the 3D structure of the GRP75 protein need to be further investigated. Alternatively, p-S148 GRP75 may provide binding sites for binding partners in MAMs or in the cytosol, which may also prevent its translocation to the mitochondria. Notably, recent work by Zhang and colleagues has shown that MAMs plays essential roles in ferroptosis [50]. It warrants further exploration whether p-S148 GRP75 influence MAM formation and thus ferroptosis.
此外,MAM 上 GRP75 的一个已知功能是通过与 OMM 中的 VDAC1 和 ER 膜上的 IP3R 相互作用来束缚 ER 和线粒体 [ 47 ]。一些调节因子,如 TG2 [ 48 ] 和 DJ-1 [ 49 ] 已被确定可通过与 GRP75 相互作用来调节 MAM 的形成。最近,PDK4 介导的 GRP75 磷酸化也被证明可以促进酒精相关性肝病中 MAM 的形成 [ 38 ]。我们的数据表明,PKA 诱导的铁死亡诱导的 GRP75 磷酸化导致 GRP75 被锁定在线粒体之外。在 GRP75 蛋白的一级序列中,残基 S148 远离线粒体定位信号(1-46 个氨基酸,aa)。它们在 GRP75 蛋白 3D 结构中的相对空间位置需要进一步研究。或者,p-S148 GRP75 可以为 MAM 或胞质溶胶中的结合配偶体提供结合位点,这也可以防止其易位到线粒体。值得注意的是,Zhang 及其同事最近的工作表明,MAM 在铁死亡中发挥着重要作用 [ 50 ]。值得进一步探索 p-S148 GRP75 是否影响 MAM 形成并从而影响铁死亡。
Furthermore, a number of conventional oncogenes or tumor suppressors have been reported to inhibit cancer development through mechanisms not previously implicated in the regulation of ferroptosis [10, 51,52,53]. We found that GRP75 KO improved the responses of cancer cells to ferroptosis both in vitro and in vivo. Importantly, WT GRP75 abolished the effect of GRP75 KO, whereas S148A did not, suggesting that PKA-mediated GRP75 phosphorylation is essential for its antiferroptotic activity. This is interesting, because a number of GRP75 inhibitors are in various stages of preclinical and clinical testing [54,55,56]. However, none have been approved by the FDA to date. It will be interesting to develop an alternative GRP75 inhibitory strategy by targeting S148, which may be effective at least when combined with pro-ferroptosis treatment.
此外,据报道,许多传统的癌基因或肿瘤抑制基因可以通过以前未涉及铁死亡调节的机制来抑制癌症的发展[10,51,52,53 ] 。我们发现 GRP75 KO 在体外和体内均改善了癌细胞对铁死亡的反应。重要的是,WT GRP75消除了GRP75 KO的作用,而S148A则没有,这表明PKA介导的GRP75磷酸化对其抗铁死亡活性至关重要。这很有趣,因为许多 GRP75 抑制剂正处于临床前和临床测试的不同阶段 [54,55,56 ] 。然而,迄今为止,尚未获得 FDA 的批准。通过针对 S148 开发一种替代的 GRP75 抑制策略将会很有趣,该策略至少在与促铁死亡治疗相结合时可能有效。
Another finding of this study is that, once GRP75 is sequestered outside the mitochondria, it promotes the stability of Nrf2 by interfering with the interaction between Nrf2 and its primary inhibitor Keap1, which is mainly localized in the cytosol. Nrf2 is a well-known antioxidant transcription factor [57]. We recently demonstrated that Nrf2 also transcriptionally activates the expression of proinflammatory genes in a ROS-independent manner [58]. With the identification of the role of Nrf2 in ferroptosis, its activity in the regulation of iron and GSH metabolism genes has been also reported [43, 59]. Our data show that GRP75 is broadly involved in various ferroptosis-related pathways, which supports the finding that GRP75 acts mainly by activating Nrf2. In addition, although GRP75 contains both low affinity DLG and high affinity ETGE motifs, only ETGE is exposed on protein surface and therefore contributes to the binding with Keap1. It has been shown previously that both DLG and ETGE are required for Keap1-mediated Nrf2 degradation [60]. This finding provides an explanation why GRP75 binds with Keap1, but is not able to degraded by Keap1. It should be noted that Keap1 is a master regulator of Nrf2. Our data show that GRP75 inhibits ferroptosis mainly by interfering with the interaction between Keap1 and Nrf2. Although additional factors such as BACH1 have been shown to regulate Nrf2 independently of Keap1 [61]. Its contribution is unlikely to be significant in our experimental settings.
这项研究的另一个发现是,一旦 GRP75 被隔离在线粒体外,它就会通过干扰 Nrf2 与其主要抑制剂 Keap1(主要位于细胞质中)之间的相互作用来促进 Nrf2 的稳定性。 Nrf2 是一种众所周知的抗氧化转录因子[ 57 ]。我们最近证明,Nrf2 还以不依赖于 ROS 的方式转录激活促炎基因的表达 [ 58 ]。随着 Nrf2 在铁死亡中的作用的确定,其在铁和 GSH 代谢基因调节中的活性也已被报道 [ 43 , 59 ]。我们的数据显示GRP75广泛参与各种铁死亡相关途径,这支持了GRP75主要通过激活Nrf2发挥作用的发现。此外,虽然GRP75同时含有低亲和力DLG和高亲和力ETGE基序,但只有ETGE暴露在蛋白质表面,因此有助于与Keap1的结合。先前已表明,DLG 和 ETGE 都是 Keap1 介导的 Nrf2 降解所必需的 [ 60 ]。这一发现解释了为什么 GRP75 与 Keap1 结合,但不能被 Keap1 降解。值得注意的是,Keap1 是 Nrf2 的主调节器。我们的数据表明,GRP75 主要通过干扰 Keap1 和 Nrf2 之间的相互作用来抑制铁死亡。尽管 BACH1 等其他因素已被证明可以独立于 Keap1 调节 Nrf2 [ 61 ]。在我们的实验环境中,它的贡献不太可能显着。
In conclusion, we have identified a localized signaling cascade (AKAP1/PKA/GRP75) involved in the protection of cells from ferroptosis, which broadens our understanding of how cells defend themselves against ferroptosis and also provides a new target that may improve the responses of cancer cells to ferroptosis.
总之,我们发现了一个参与保护细胞免受铁死亡的局部信号级联(AKAP1/PKA/GRP75),这拓宽了我们对细胞如何防御铁死亡的理解,并提供了一个可能改善癌症反应的新靶点细胞铁死亡。
Methods and Details 方法和细节
Colon cancer patient samples
结肠癌患者样本
Forty pairs of colon cancer tissues and their adjacent normal controls were collected from the Third Affiliated Hospital of Harbin Medical University, China. All patients received written informed consent. The samples were collected immediately after surgery and stored in liquid nitrogen for future analysis. Total protein was extracted for western blot assay. This study has been approved by the Research Ethics Committee of Harbin Medical University, China.
四十对结肠癌组织及其癌旁正常对照取自哈尔滨医科大学附属第三医院。所有患者均获得书面知情同意书。手术后立即收集样本并储存在液氮中以供将来分析。提取总蛋白用于蛋白质印迹测定。本研究已获得中国哈尔滨医科大学研究伦理委员会的批准。
Cell lines and treatments
细胞系和治疗
The human colon cancer cell lines HCT116, LoVo and HT-29, human cervical cancer cell line HeLa, human liver cancer cell line 97H and human osteosarcoma cell line HT1080 were maintained at 10% (v/v) fetal bovine serum (FBS) in RPMI-1640 medium (Gibco) or DMEM medium (Gibco). All cells were cultured in a humidified incubator (Thermo Scientific) containing 5% CO2 at 37 °C. The cell lines were routinely tested to rule out mycoplasma contamination and authenticated using short tandem repeat markers by Gene Detection Biotechnology (Suzhou, China). All siRNA oligonucleotides and plasmids were transfected with commercial Lipofectamine 2000 (Invitrogen) according to product instructions. The indicated siRNA sequences were listed in Supplemental Table 1. 2 mM sulfasalazine (SAS), 5 μM Erastin, 5 μM ML162, 3 μM RSL-3 and DMSO were used to treat cells for 24 h to induce ferroptosis. The sequences of siRNA were listed in Supplemental Table 1.
将人结肠癌细胞系 HCT116、LoVo 和 HT-29、人宫颈癌细胞系 HeLa、人肝癌细胞系 97H 和人骨肉瘤细胞系 HT1080 维持在 10% (v/v) 胎牛血清 (FBS) 中。 RPMI-1640 培养基 (Gibco) 或 DMEM 培养基 (Gibco)。所有细胞均在含有5% CO 2 的加湿培养箱(Thermo Scientific)中于37℃培养。对细胞系进行常规测试以排除支原体污染,并使用基因检测生物技术公司(中国苏州)的短串联重复标记进行验证。根据产品说明,用商业 Lipofectamine 2000 (Invitrogen) 转染所有 siRNA 寡核苷酸和质粒。补充表1中列出了指定的 siRNA 序列。使用 2 mM 柳氮磺吡啶 (SAS)、5 μM Erastin、5 μM ML162、3 μM RSL-3 和 DMSO 处理细胞 24 小时以诱导铁死亡。 siRNA的序列列于补充表1中。
Western blot 蛋白质印迹
Cells and tumor tissues were lyzed in urea buffers containing 2 m thiourea, 4% CHAPS, 40 mM Tris-Base, 40 mM DTT, 2% Pharmalyte. Proteins were separated by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE) and then transferred to the solid membrane for the sequential incubation with primary and secondary antibodies. The protein expression levels were determined by electrochemiluminescence (ECL) and visualized by Image studio system (ECL, LI-COR, Lincoln, Georgia). The bands were quantified by Image J software (National Institutes of Health, Bethesda, MD). The sources and dilution ratios of primary antibodies were as follows: anti-GRP75 (Santa, #A4605, 1:1000)/(Proteintech, #10503-2-AP, 1:1000), anti-P-S/T(Abclonal, #ab1101, 1:1000), anti-CNX (Proteintech, #12987-1-AP, 1:2000), anti-Keap1 (Proteintech, #10503-2-AP, 1:2000), anti-AKAP-1 (Proteintech, #12489-1-AP, 1:1000), anti-β-actin (Abclonal, #AC004, 1:2000), anti-VDAC1 (Proteintech, #55259-1-AP, 1:2000), anti-α-tubulin (Proteintech, #11224-1-AP, 1:2000), anti-HSP60 (Abclonal, #A0969, 1:2000), anti-Nrf2 (Proteintech, #16396-1-AP, 1:1000), anti-FLAG-tag (Proteintech, #66008-4-Ig, 1:1000)/(Proteintech, #80010-1-RR, 1:1000), anti-IgG-Rabbit (ABclonal, #7016, 1:100), anti-IgG-Mouse (Beyotime, #7028, 1:100), anti-PKA-Cα (CST, #4782, 1:1000), anti-HA-tag (CST, #3724, 1:1000), anti-GST (ABclonal, #AE006, 1:2000)/(Proteintech, #HRP-66001, 1:2000), anti-FSP1 (Santa, #sc-377120, 1:1000), anti-FTH (Proteintech, #11682-1-AP, 1:1000), anti-GPX4 (Abcam, #125066, 1:1000).
细胞和肿瘤组织在含有 2 m 硫脲、4% CHAPS、40 mM Tris-Base、40 mM DTT、2% Pharmalyte 的尿素缓冲液中裂解。通过十二烷基硫酸钠 (SDS)-聚丙烯酰胺凝胶电泳 (PAGE) 分离蛋白质,然后转移到固体膜上,与一抗和二抗依次孵育。通过电化学发光 (ECL) 测定蛋白质表达水平,并通过 Image studio 系统 (ECL, LI-COR, Lincoln, Georgia) 进行可视化。通过 Image J 软件(美国国立卫生研究院,贝塞斯达,马里兰州)对条带进行量化。一抗来源及稀释比例如下:抗GRP75(Santa, #A4605, 1:1000)/(Proteintech, #10503-2-AP, 1:1000)、抗PS/T(Abclonal, # ab1101, 1:1000), 抗 CNX (Proteintech, #12987-1-AP, 1:2000)、抗 Keap1 (Proteintech, #10503-2-AP, 1:2000)、抗 AKAP-1 (Proteintech, #12489-1-AP, 1:1000)、抗 β-肌动蛋白 (Abclonal , #AC004, 1:2000), 抗 VDAC1 (Proteintech, #55259-1-AP, 1:2000)、抗 α-微管蛋白 (Proteintech, #11224-1-AP, 1:2000)、抗 HSP60 (Abclonal, #A0969, 1:2000)、抗 Nrf2 (蛋白质技术,#16396-1-AP,1:1000),抗 FLAG 标签(Proteintech,#66008-4-Ig,1:1000)/(Proteintech,#80010-1-RR,1:1000),抗 IgG-Rabbit(ABclonal,#7016,1:100) , 抗 IgG-Mouse (Beyotime, #7028, 1:100), 抗 PKA-Cα (CST, #4782, 1:1000)、抗 HA 标签 (CST, #3724, 1:1000)、抗 GST (ABclonal, #AE006, 1:2000)/(Proteintech, #HRP-66001, 1 :2000),抗 FSP1(圣诞老人,#sc-377120, 1:1000)、抗 FTH (Proteintech, #11682-1-AP, 1:1000)、抗 GPX4 (Abcam, #125066, 1:1000)。
RNA extraction and quantitative (q)RT-PCR
RNA 提取和定量 (q)RT-PCR
RNA was extracted using an RNA extraction kit (TaKaRa) according to the experimental procedure provided by the RNA extraction kit. The isolated total RNA was retrotranscribed using Abclonal reverse transcriptional system kit. qRT-PCR was performed by using SYBR Premix Ex Tag II (TaKaRa). The expression of genes relative to the 18 s rRNA control was calculated by 2-△△ct. The sequences of primer were listed in Supplementary Table 1.
根据RNA提取试剂盒提供的实验步骤,使用RNA提取试剂盒(TaKaRa)提取RNA。使用 Abclonal 逆转录系统试剂盒对分离的总 RNA 进行逆转录。使用SYBR Premix Ex Tag II (TaKaRa)进行qRT-PCR。通过2-△△ ct计算相对于18s rRNA对照的基因表达量。引物序列列于补充表1中。
Luciferase reporter assay
荧光素酶报告基因检测
Equal amounts of Nrf2-antioxidant response element (ARE)-luciferase reporter plasmids were transfected into indicated in the control and GRP75 KD cells. Each transfection contained equal amounts of Renilla plasmid for standardized control. 24 h after transfection, the cells were treated with SAS(2 mM) or DMSO for 24 h. Luciferase activity was measured using the luciferase assay system kit (Promega). The relative luciferase activity was normalized with the Leighton luciferase activity.
将等量的 Nrf2-抗氧化反应元件 (ARE)-荧光素酶报告质粒转染至对照和 GRP75 KD 细胞中。每次转染均含有等量的海肾质粒以进行标准化对照。转染24小时后,用SAS(2mM)或DMSO处理细胞24小时。使用荧光素酶测定系统试剂盒(Promega)测量荧光素酶活性。相对荧光素酶活性用 Leighton 荧光素酶活性标准化。
Immunoprecipitation (IP) 免疫沉淀 (IP)
The treated cells were lysed in the NETN buffer (50 mM Tris-HCl [pH=8.0], 150 mM NaCl, 1% NP-40, 1 mM EDTA) supplemented with Protease Inhibitor Cocktail (1:100) (MedChemExpress, #HY-K0010). After pre-cleaning with protein G sepharose beads 4 Fast Flow (GE Healthcare, #17061802), specific antibodies or IgG control were added to the supernatant and incubated with FBS blocked beads on a runner at 4 °C for about 20 h. The beads bound to the immunoprecipitates were washed three times with NETN buffer, and then the samples were detected by western blot assay. And the bands were quantified by Image J software (National Institutes of Health, Bethesda, MD).
将处理过的细胞在补充有蛋白酶抑制剂混合物 (1:100) (MedChemExpress, #HY) 的 NETN 缓冲液(50 mM Tris-HCl [pH=8.0]、150 mM NaCl、1% NP-40、1 mM EDTA)中裂解。 -K0010)。用 Protein G Sepharose Bead 4 Fast Flow(GE Healthcare,#17061802)预清洗后,将特异性抗体或 IgG 对照添加到上清液中,并与 FBS 封闭的珠子在转轮上于 4 °C 下孵育约 20 小时。将与免疫沉淀物结合的珠子用NETN缓冲液洗涤3次,然后通过蛋白质印迹法检测样品。并且通过 Image J 软件(美国国立卫生研究院,贝塞斯达,马里兰州)对条带进行量化。
Immunohistochemistry 免疫组织化学
Different groups of xenograft sections were fixed by paraffin, and then sequentially treated with xylene and different concentrations of alcohol solutions to remove paraffin. Then these sections were subjected to antigen retrieval by boiling in 0.01 mol/L citrate buffer for 15 min. After cooling to room temperature, all tissue sections were incubated at 4 °C overnight with anti-Ki67 (Proteintech, #27309-1-AP, 1:1000) antibodies and anti-4-HNE (Abcam, ab46545, 1:400) antibodies. 3,3’Diaminobenzidine (DAB) peroxidase substrate kit (ZSBG-BIO) was used to visualize the signal. 5 images were randomly taken for each slice and saved. The protein expression effencity was quantified by Image J software (National Institutes of Health, Bethesda, MD).
将不同组的异种移植切片用石蜡固定,然后依次用二甲苯和不同浓度的酒精溶液处理以除去石蜡。然后将这些切片在0.01 mol/L柠檬酸盐缓冲液中煮沸15分钟进行抗原修复。冷却至室温后,将所有组织切片与抗 Ki67(Proteintech,#27309-1-AP,1:1000)抗体和抗 4-HNE(Abcam,ab46545,1:400)在 4 °C 下孵育过夜抗体。使用 3,3' 二氨基联苯胺 (DAB) 过氧化物酶底物试剂盒 (ZSBG-BIO) 使信号可视化。每个切片随机拍摄 5 张图像并保存。通过 Image J 软件(美国国立卫生研究院,贝塞斯达,马里兰州)对蛋白质表达效率进行量化。
Purification of recombinant protein
重组蛋白的纯化
The cDNA sequences of the genes used in the experiments were cloned into pGEX-6P-1 expression vector. After introduced the plasmids into Escherichia coli ER2566, the expression of the corresponding protein was induced by adding 0.1 mM isopropyl β-D-1-thiogalactoside (IPTG) in the cultured bacteria for about 20 h at 16 °C. The recombinant proteins were purified by BeyoGold™GST-tag purification resin (Beyotime) and collected using GSH eluting buffers. The expression efficiency of the above recombinant proteins was detected by western blot assay.
将实验中使用的基因的cDNA序列克隆到pGEX-6P-1表达载体中。将质粒导入大肠杆菌ER2566后,在培养菌中添加0.1 mM异丙基β-D-1-硫代半乳糖苷(IPTG),16℃培养约20 h,诱导相应蛋白的表达。重组蛋白通过 BeyoGold™ GST 标签纯化树脂 (Beyotime) 纯化,并使用 GSH 洗脱缓冲液收集。采用western blot检测上述重组蛋白的表达效率。
Ubiquitination assay 泛素化检测
The Ub-HA plasmid and siRNA (two independent si-GRP75 oligos) were transfected into cells. The cells were treated with 20 µM MG132 (MedChemExpress, #HY-K0010) and/or DMSO, SAS (2 mM) for 6 h prior to harvest. The cells were lysed in lysis buffer containing 2% SDS, 150 mM NaCl, 10 mM Tris-HCl [pH=8.0] freshly supplemented with Protease Inhibitor Cocktail (1:100) (MedChemExpress). The resulting lysates were subjected to the IP of Nrf2 and western blot assay of anti-HA. The protein expression levels were determined by electrochemiluminescence (ECL) and visualized by Image studio system (ECL, LI-COR, Lincoln, Georgia). The bands were quantified by Image J software (National Institutes of Health, Bethesda, MD).
将 Ub-HA 质粒和 siRNA(两个独立的 si-GRP75 寡核苷酸)转染至细胞中。收获前用 20 µM MG132(MedChemExpress,#HY-K0010)和/或 DMSO、SAS (2 mM) 处理细胞 6 小时。将细胞在含有 2% SDS、150 mM NaCl、10 mM Tris-HCl [pH=8.0] 的裂解缓冲液中裂解,并新鲜补充蛋白酶抑制剂混合物 (1:100) (MedChemExpress)。将所得裂解物进行 Nrf2 的 IP 和抗 HA 的蛋白质印迹测定。通过电化学发光 (ECL) 测定蛋白质表达水平,并通过 Image studio 系统 (ECL, LI-COR, Lincoln, Georgia) 进行可视化。通过 Image J 软件(美国国立卫生研究院,贝塞斯达,马里兰州)对条带进行量化。
Proximity Ligation Assay (PLA)
邻近连接分析 (PLA)
The PLA signal is determined using Duolink® reagent (Sigma-Aldrich) according to the experimental operating instructions of the PLA assay kit. Briefly, the cells were fixed with 4% paraformaldehyde at room temperature, and then permeabilized with 0.1% Triton-X100 at 4 °C for 3 min, followed by blockade in blocking buffer at 37 °C for 1 h. Cells were incubated overnight with theindicated primary antibodies at 4 °C. After three washes in Wash buffer A, 5 min each, the slides were incubated with the PLA PLUS and MINUS secondary probes for 1 h at 37 °C, followed by the incubation with Duolink ligation solution for 30 min at 37 °C. The amplified Duolink mixture was added and incubated with slides in dark for 1.5 h at 37 °C. After washes, the nucleus was stained with DAPI, and the slide was sealed and dried overnight in dark. The signal was amplified by ligation. The images were photographed using a confocal microscope (LSM880, Zeiss).
根据 PLA 检测试剂盒的实验操作说明,使用 Duolink® 试剂 (Sigma-Aldrich) 测定 PLA 信号。简而言之,细胞在室温下用4%多聚甲醛固定,然后用0.1% Triton-X100在4℃下透化3分钟,然后在封闭缓冲液中在37℃下封闭1小时。将细胞与指定的一抗在 4°C 下孵育过夜。在洗涤缓冲液 A 中洗涤 3 次(每次 5 分钟)后,将载玻片与 PLA PLUS 和 MINUS 二级探针在 37 °C 下孵育 1 小时,然后与 Duolink 连接溶液在 37 °C 下孵育 30 分钟。添加扩增的 Duolink 混合物,并在 37°C 下与载玻片在黑暗中孵育 1.5 小时。洗涤后,用DAPI对细胞核进行染色,并将载玻片密封并在黑暗中干燥过夜。通过连接放大信号。使用共焦显微镜(LSM880,Zeiss)拍摄图像。
CCK8 assay CCK8检测
Cell viability was detected according to the experimental instructions of Cell Counting Kit (SEVEN). The concentrated detection solution was mixed with serum-free medium at a ratio of 1:9. Add 100 μL of detection buffer to each test hole. After incubation at 37 °C for about 2 h in dark, the absorbance value at 450 nm was detected using Microplate Reader (Tecan Austria GmbH 5082, Grodig).
按照细胞计数试剂盒(SEVEN)实验说明检测细胞活力。将浓缩的检测液与无血清培养基以1:9的比例混合。每个测试孔中添加 100 μL 检测缓冲液。 37℃避光孵育约2小时后,使用酶标仪(Tecan Austria GmbH 5082,Grodig)检测450 nm处的吸光度值。
LDH release assay LDH释放测定
Cell death rate was measured according to the instructions of the lactate dehydrogenase cytotoxicity assay kit (Beyotime). First, the LDH detection working solution was configured with a ratio of 91 μL of detection buffer to 9 μL of chromogen solution and incubated at room temperature without light for 20 min. After the addition of 20 µl termination solution, the absorbance value at 450 nm was detected using Microplate Reader (Tecan Austria GmbH 5082, Grodig).
按照乳酸脱氢酶细胞毒性测定试剂盒(Beyotime)说明书测定细胞死亡率。首先按照91μL检测缓冲液与9μL显色液的比例配置LDH检测工作液,室温避光孵育20min。添加20 µl终止液后,使用酶标仪(Tecan Austria GmbH 5082,Grodig)检测450 nm处的吸光度值。
PKA kinase activity assay
PKA激酶活性测定
Cells were collected and the PKA activity in cells were detected following the instructions of the PKA kinase activity detection kit (Enzo). Simply put, 30 μL of cell lysate is added to the microdrop plate. 10 μL of diluted 1 mg/mL ATP to each well, and incubated at 30 °C for 1.5 h. Remove the solution from the detector hole and add 40 μL of Phosphospecific Substrate Antibody, and incubate at room temperature for 1 h. At room temperature, wash 4 times with 1 x wash buffer for 5 min each time. Add 40 μL of diluted Anti-Rabbit IgG HRP Conjugate to each well, and incubated at room temperature for 1.5 h. Wash 4 times with 1 x washing buffer for 5 min each time. Add 60 μL of TMB Substrate to each well, incubated at room temperature for 1.5 h. Add 20 μL of stop solution 2 to each well. Finally, the absorbance of the reaction mixture was detected at 450 nm using a Microplate Reader (Tecan Austria GmbH 5082, Grodig).
收集细胞,按照PKA激酶活性检测试剂盒(Enzo)说明书检测细胞内PKA活性。简单地说,将 30 μL 细胞裂解液添加到微滴板中。每孔加入 10 μL 稀释的 1 mg/mL ATP,30℃孵育 1.5 h。从检测器孔中取出溶液,加入 40 μL 磷酸化底物抗体,室温孵育 1 h。室温下,用 1 x 洗涤缓冲液洗涤 4 次,每次 5 分钟。每孔加入 40 μL 稀释的抗兔 IgG HRP 结合物,室温孵育 1.5 小时。用 1 x 洗涤缓冲液洗涤 4 次,每次 5 分钟。每孔加入 60 μL TMB 底物,室温孵育 1.5 h。每孔加入 20 μL 终止液 2。最后,使用酶标仪(Tecan Austria GmbH 5082,Grodig)在 450 nm 处检测反应混合物的吸光度。
In vitro kinase assay 体外激酶测定
The experiments were performed following the instruction provided by the PKA-catalytic subunit purified protein kit (New England Biolabs). Briefly, in vitro purified WT GRP75 and GRP75 S148A proteins were respectively added to the reaction system containing PKACA kinase, NEBuffer™ and 500 μM ATP. The mixtures were incubated at 30 °C for 1 h. After that, SDS loading buffer was added to the mixture to terminate the reaction. The phosphorylation signal was detected by western blot of Anti-Phospho - (Ser/Thr) antibody.
实验按照 PKA 催化亚基纯化蛋白试剂盒(New England Biolabs)提供的说明进行。简而言之,将体外纯化的WT GRP75和GRP75 S148A蛋白分别添加到含有PKAKA激酶、NEBuffer™和500μM ATP的反应体系中。将混合物在 30°C 下孵育 1 小时。之后,将SDS加样缓冲液添加至混合物中以终止反应。通过抗磷酸-(Ser/Thr)抗体的蛋白质印迹检测磷酸化信号。
Lipid ROS assay 脂质ROS测定
C11 BODIPY 581/591 (invitrogen) is a lipid soluble fluorescent probe used to detect lipid peroxidation levels in living cells. The probe was dissolved in the medium without serum (1:1000) and incubated at 37 °C for 30 min without light. Then, the PBS was washed three times and the single-cell suspension was prepared. The cells incubated with the probe were detected by flow cytometry at 488 nm and Lipid ROS levels were calculated. The distribution of lipid ROS in cells were visualized by SIM microscopy (Beijing Nanolnsights-tech Co., Ltd.).
C11 BODIPY 581/591 (invitrogen) 是一种脂溶性荧光探针,用于检测活细胞中的脂质过氧化水平。将探针溶解在无血清培养基(1:1000)中,37℃避光孵育30分钟。然后用PBS洗涤3次,制备单细胞悬液。通过流式细胞仪在 488 nm 处检测与探针孵育的细胞,并计算脂质 ROS 水平。通过SIM显微镜(北京纳创科技有限公司)观察细胞内脂质ROS的分布。
Subcellular fractionation
亚细胞分离
Subcellular fractionation was performed as previously described [62]. Briefly, cells (2 × 109) pellet were resuspended in IBcells-1 buffers (225 mM mannitol, 75 mM sucrose, 0.1 mM EGTA and 30 mM Tris-HCl [pH7.4]) in ice bath and gently disrupted by repeated flaps in Dounce homogenization. Cell lysates were centrifuged at 600 g for 5 min at 4 °C to remove the nucleus and unbroken cells. Then the supernatant was collected, and subjected to another round of centrifuged at 7000 g for 20 min to separate crude mitochondria (pellet) and others (supernatant). Further centrifugation of the supernatant at 100,000 g for 90 min at 4 °C to obtain the ER fraction (pellet) and cytoplasmic fraction (supernatant). The crude mitochondrial fraction was resuspended in 2 mL mitochondria resuspending buffer (MRB, 250 mM mannitol, 5 mM HEPES pH 7.4 and 0.5 mM EGTA). The suspension was layered on top of 30% percoll medium (225 mM mannitol, 25 mM HEPES [pH7.4], 1 mM EGTA and 30% vol/vol Percoll) and centrifuged at 95,000 g for 30 min at 4 °C to obtain MAMs and pure mitochondria fractions. The MAMs fraction was further centrifuged to remove the contaminated mitochondria. All fractions are collected to further assays.
如前所述进行亚细胞分级分离[ 62 ]。简而言之,将细胞 (2 × 10 9 ) 沉淀重悬于冰浴中的 IBcells-1 缓冲液(225 mM 甘露醇、75 mM 蔗糖、0.1 mM EGTA 和 30 mM Tris-HCl [pH7.4])中,并通过重复瓣膜轻轻破碎在杜恩斯同质化中。将细胞裂解物在 4°C 下以 600 g 离心 5 分钟,以去除细胞核和未破碎的细胞。然后收集上清液,再进行一轮7000 g离心20 min,分离粗线粒体(沉淀)和其他(上清液)。将上清液在 4℃、100,000 g 下进一步离心 90 分钟,以获得 ER 级分(沉淀)和细胞质级分(上清液)。将粗线粒体部分重悬于 2 mL 线粒体重悬缓冲液(MRB、250 mM 甘露醇、5 mM HEPES pH 7.4 和 0.5 mM EGTA)中。将悬浮液分层在 30% Percoll 培养基(225 mM 甘露醇、25 mM HEPES [pH7.4]、1 mM EGTA 和 30% vol/vol Percoll)上,并在 4 °C 下以 95,000 g 离心 30 分钟,以获得MAM 和纯线粒体组分。 MAM 部分进一步离心以去除污染的线粒体。收集所有级分以进行进一步测定。
Digitonin can selectively penetrate the plasma membrane rather than the organelle membrane, and therefore used to analysis the proteins that released from membrane organelles [63, 64]. As such, cells were collected after the indicated treatments. One third cells were lyzed with Urea buffer as described above to obtain total cell lysates. Another 2/3 of the cells were re-suspended in a transport buffer (20 mM HEPES [pH=7.4], 110 mM potassium acetate, 2 mM magnesium acetate, 0.5 mM EGTA) supplemented with Protease Inhibitor Cocktail (1:100) (MedChemExpress, #HY-K0010). The cell permeability was performed by adding 0.01% digitalin into the cell suspension on iced for 5 min. After centrifugation at 1000 g at 4 °C, the precipitation (permeabilized cell fraction) and supernatant (released fraction) were collected for further assay. The samples were detected by western blot assay. And the bands were quantified by Image J software (National Institutes of Health, Bethesda, MD).
洋地黄皂苷可以选择性地穿透质膜而不是细胞器膜,因此可用于分析从膜细胞器释放的蛋白质[ 63 , 64 ]。因此,在指定的处理后收集细胞。如上所述用尿素缓冲液裂解三分之一细胞以获得总细胞裂解物。另外 2/3 的细胞重悬于补充有蛋白酶抑制剂混合物 (1:100) 的运输缓冲液(20 mM HEPES [pH=7.4]、110 mM 乙酸钾、2 mM 乙酸镁、0.5 mM EGTA)中( MedChemExpress,#HY-K0010)。通过将0.01%洋地黄素添加到冰上的细胞悬液中5分钟来检测细胞通透性。 4°C 1000 g 离心后,收集沉淀(透化细胞级分)和上清液(释放级分)用于进一步测定。通过蛋白质印迹法检测样品。并且通过 Image J 软件(美国国立卫生研究院,贝塞斯达,马里兰州)对条带进行量化。
In vivo xenograft mouse study
小鼠体内异种移植研究
Four-to-five week old female nude mice were purchased from Liaoning Changsheng Biological Co., LTD for the subsequent assays. First, GRP75 KO stable HCT116 cell line was infected with lentivirus expressing the vector control, WT GRP75 or GRP75 S148A to establish the sublines with different genetic background of GRP75. The expression levels of GRP75 were determined by western blot assay. After that, the same number of HCT116 sublines (1 × 107) were inoculated subcutaneously on the dorsal side (both sides) of the mice. When the tumor volumes reached about 200 mm3, HCT116/control (Ctl.), GRP75 KO, GRP75 KO + WT GRP75 and GRP75 KO + GRP75 S148A bearing mice, each of which were randomly divided into two groups (n = 5/group), were subjected to the indicated treatments (SAS, 250 mg/kg intraperitoneal injection daily with DMSO as controls).
4-5周龄雌性裸鼠购自辽宁昌盛生物有限公司用于后续检测。首先,用表达载体对照、WT GRP75或GRP75 S148A的慢病毒感染GRP75 KO稳定HCT116细胞系,建立具有不同GRP75遗传背景的亚系。通过蛋白质印迹法测定GRP75的表达水平。之后,在小鼠背侧(两侧)皮下接种相同数量的HCT116亚系(1×10 7 )。当肿瘤体积达到约200mm 3 时,将HCT116/control(Ctl.)、GRP75KO、GRP75KO+WTGRP75和GRP75KO+GRP75S148A荷瘤小鼠随机分为两组( n =5/组) ),接受指定的治疗(SAS,每天腹腔注射 250 mg/kg,用 DMSO 作为对照)。
For the patient-derived xenograft (PDX) assay, human colon cancer tissue was sliced into small pieces of 2–3 mm and transplanted under sterile conditions into 4–5 week-old female NOD-SCID IL2RGamma null (NOD-SCID Gamma, NSG) mice (Changsheng Biology). After 3 passages of PDX in NSG mice, the xenografts bearing mice were used for follow-up drug treatment. When the tumor grew to about 150 mm3, they were randomly divided into 5 groups: (i) DMSO (intraperitoneal injection daily), (ii) SAS (250 mg/kg intraperitoneal injection daily), (iii) MKT077 (10 mg/kg intraperitoneal injection, once every two days), (iv) SAS combined with MKT077, (v) SAS combined with MKT077 and Lip-1. After the administration of the vehicle, SAS and MKT077 for 20 days, we stopped the treatments and analyzed the tumors size, tumor weight and body weight.
对于患者来源的异种移植 (PDX) 测定,将人结肠癌组织切成 2-3 毫米的小块,并在无菌条件下移植到 4-5 周龄雌性 NOD-SCID IL2RGamma null 中(NOD-SCID Gamma,NSG) )小鼠(长生生物)。 NSG小鼠PDX传代3次后,携带异种移植瘤的小鼠用于后续药物治疗。当肿瘤生长至约150 mm 3 时,将其随机分为5组:(i)DMSO(每日腹腔注射),(ii)SAS(每日250 mg/kg腹腔注射),(iii)MKT077(10 mg/kg) kg腹腔注射,每两天一次),(iv)SAS联合MKT077,(v)SAS联合MKT077和唇-1。给予媒介物、SAS和MKT077 20天后,我们停止治疗并分析肿瘤大小、肿瘤重量和体重。
All animal studies were conducted in accordance with the National Institute of Health guidelines for the Care and Use of Laboratory Animals and approved by the Ethics Committee of Harbin Medical University Cancer Hospital.
所有动物研究均按照美国国立卫生研究院实验动物护理和使用指南进行,并经哈尔滨医科大学肿瘤医院伦理委员会批准。
Liquid Chromatography Mass Spectrometry (LC-MS) analysis
液相色谱质谱 (LC-MS) 分析
Briefly, the cells treated with DMSO or SAS (2 mM, 24 h) were subjected to IP of GRP75. The eluted proteins from IP were concentrated with SDS-PAGE gel. The gel bands were dehydrated, reduced, alkylated and digested with trypsin at 37 °C for 16 h. After digestion, the peptides were extracted with acetonitrile (ACN), dried using vacuum, and then dissolved in 0.1% formic acid (FA) with 2% ACN. The peptides were separated onto a nano RP column (1.7 µm, 150 µm x 25 cm, GmbH) and eluted with a gradient (5–35%) of ACN for 45 min at 500 nL/min. The separated peptides were subjected to Orbitrap Fusion Lumos (Thermo Fisher Scientific, San Jose, CA) mass spectrometry in DDA (data-dependent Acquisition) mode, with a full MS scan from 350 to 1250 m/z and top 20 precursors selected for fragmentation. Raw data was further analyzed using Maxquant v2.4.3.0 [65] against the human SwissProt_new xyzzy database (20 255 sequences) with 1% FDR at protein level.
简而言之,用 DMSO 或 SAS(2 mM,24 小时)处理的细胞进行 GRP75 的 IP。用 SDS-PAGE 凝胶浓缩 IP 洗脱的蛋白质。将凝胶条带脱水、还原、烷基化并用胰蛋白酶在 37°C 下消化 16 小时。消化后,用乙腈 (ACN) 提取肽,真空干燥,然后溶解在含 2% ACN 的 0.1% 甲酸 (FA) 中。将肽分离到纳米 RP 柱(1.7 µm,150 µm x 25 cm,GmbH)上,并用乙腈梯度 (5–35%) 以 500 nL/min 的速度洗脱 45 分钟。分离的肽在 DDA(数据依赖采集)模式下进行 Orbitrap Fusion Lumos(Thermo Fisher Scientific,圣何塞,加利福尼亚州)质谱分析,从 350 到 1250 m/z 进行全 MS 扫描,并选择前 20 个前体进行裂解。使用 Maxquant v2.4.3.0 [ 65 ] 针对人类 SwissProt_new xyzzy 数据库(20 255 个序列)进一步分析原始数据,蛋白质水平的 FDR 为 1%。
Detection of cAMP levels cAMP水平检测
cAMP levels were determined according to the instructions provided by the manufacturer of the human cAMP ELISA Kit (COIBO BIO, CB10703-Hu). Briefly, 50 µl lysate derived from the indicated cells was added to 96 microwell plates pre-coated with cAMP antibody, and then 100 µl of HRP labeled detection antibody was added, and the mixtures were incubate at 37 °C for 60 min. Then, discarded the liquid and followed by five washes in 1 x washing buffer for 1 min each. 50 µl of chromogen solution A and chromogen solution B were added to each well, and incubated with the mixture at 37 °C for 15 min in dark. Finally, 50 µl stop buffer was added to stop the reaction and the absorbance of the sample at 450 nm was measured by a Microplate Reader (Tecan Austria GmbH 5082, Grodig).
根据人 cAMP ELISA 试剂盒(COIBO BIO,CB10703-Hu)制造商提供的说明测定 cAMP 水平。简而言之,将 50 µl 所示细胞裂解液加入到预先包被有 cAMP 抗体的 96 微孔板中,然后加入 100 µl HRP 标记的检测抗体,并将混合物在 37°C 下孵育 60 分钟。然后,丢弃液体,然后在 1 x 洗涤缓冲液中洗涤五次,每次 1 分钟。每孔加入显色剂溶液 A 和显色剂溶液 B 50 µl,并在 37°C 下避光孵育 15 分钟。最后,添加 50 µl 终止缓冲液以终止反应,并通过酶标仪(Tecan Austria GmbH 5082,Grodig)测量样品在 450 nm 处的吸光度。
Immunofluorescence staining
免疫荧光染色
Briefly, cells were fixed in 4% paraformaldehyde for 20 min, followed by three washes in PBS and permeabilization in 0.1%Triton-X100 at 4 °C for 3 min. Cells were then blocked with 3% BSA for 1 h at room temperature and incubated with indicated primary antibody overnight at 4 °C. After that, cells were incubated with the second antibody at room temperature for 2 h in dark. Then, the nucleus was stained with DAPI, and the slide was sealed and dried overnight in dark. Images collected by the confocal microscopy (LSM880, Zeiss) with a wide-field oil objective (×63/1.3, Zeiss) were processed by the sparse deconvolution [66] to eliminate the read-out noise and enhance the contrast.
简而言之,将细胞在 4% 多聚甲醛中固定 20 分钟,然后在 PBS 中洗涤 3 次,并在 0.1% Triton-X100 中于 4 °C 透化 3 分钟。然后用 3% BSA 在室温下封闭细胞 1 小时,并与指定的一抗在 4 °C 下孵育过夜。之后,将细胞与二抗在室温下避光孵育2小时。然后,用DAPI对细胞核进行染色,将载玻片密封并在黑暗中干燥过夜。共聚焦显微镜(LSM880,Zeiss)和宽视场油物镜(×63/1.3,Zeiss)收集的图像通过稀疏反卷积[ 66 ]进行处理,以消除读出噪声并增强对比度。
Online public database 在线公共数据库
CPTAC datasets(https://ualcan.path.uab.edu/)
CPTAC 数据集( https://ualcan.path.uab.edu/ )
TCGA dataset (https://ualcan.path.uab.edu/)
TCGA 数据集 ( https://ualcan.path.uab.edu/ )
Prognostic value (http://dna00.bio.kyutech.ac.jp/)
预后价值 ( http://dna00.bio.kyutech.ac.jp/ )
TCGA database (https://portal.gdc.cancer.gov/)
TCGA 数据库 ( https://portal.gdc.cancer.gov/ )
Cancer Therapeutics Response Portal (CTRP) (https://portals.broadinstitute.org/)
癌症治疗响应门户 (CTRP) ( https://portals.broadinstitute.org/ )
Cancer Cell Line Encyclopedia (CCLE) database (https://sites.broadinstitute.org/ccle/).
癌细胞系百科全书(CCLE)数据库( https://sites.broadinstitute.org/ccle/ )。
Statistical analysis 统计分析
Statistical analysis was conducted by GraphPad software, version 8. Correlation was calculated by Pearson correlation method, and the scatter plot was draw using bioinformatics.com.cn online tool. P < 0.05 was considered statistical significance. Data are presented as the means ± standard error of the means (SEM) or standard Deviation (SD). Student t test or two-way ANOVA test was applied to assess the statistical significance. P-value < 0.05 is regarded as statistically significant.
采用GraphPad软件8版进行统计分析,采用Pearson相关法计算相关性,采用bioinformatics.com.cn在线工具绘制散点图。 P <0.05被认为具有统计学意义。数据以平均值±平均值标准误差 (SEM) 或标准偏差 (SD) 形式呈现。应用学生 t 检验或双向方差分析检验来评估统计显着性。 P值<0.05被认为具有统计显着性。
Data availability 数据可用性
Data reported in this paper will be shared by the lead contact upon request.
本文报告的数据将由主要联系人根据要求共享。
References 参考
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Dixon SJ、Lemberg KM、Lamprecht MR、Skouta R、Zaitsev EM、Gleason CE 等。铁死亡:一种铁依赖性非凋亡细胞死亡形式。细胞。 2012;149:1060–72。Zheng S, Guan XY. Ferroptosis: Promising approach for cancer and cancer immunotherapy. Cancer Lett. 2023;561:216152.
郑S,管XY。铁死亡:癌症和癌症免疫治疗的有前途的方法。癌症快报。 2023;561:216152。Stockwell BR. Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–21.
Lei G, Zhuang L, Gan B. Targeting ferroptosis as a vulnerability in cancer. Nat Rev Cancer. 2022;22:381–96.
Zhang R, Kang R, Tang D. Ferroptosis in gastrointestinal cancer: from mechanisms to implications. Cancer Lett. 2023;561:216147.
Dos Santos AF, Fazeli G, Xavier da Silva TN, Friedmann Angeli JP. Ferroptosis: mechanisms and implications for cancer development and therapy response. Trends Cell Biol. 2023;33:1062–76.
Xu H, Ye D, Ren M, Zhang H, Bi F. Ferroptosis in the tumor microenvironment: perspectives for immunotherapy. Trends Mol Med. 2021;27:856–67.
Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer. 2019;19:405–14.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell. 2017;171:273–85.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Zhao Y, Li M, Yao X, Fei Y, Lin Z, Li Z, et al. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate-Mediated AMPK-SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020;33:108487.
Tang B, Wang Y, Xu W, Zhu J, Weng Q, Chen W, et al. Macrophage xCT deficiency drives immune activation and boosts responses to immune checkpoint blockade in lung cancer. Cancer Lett. 2023;554:216021.
Kholodenko BN, Hancock JF, Kolch W. Signalling ballet in space and time. Nat Rev Mol Cell Biol. 2010;11:414–26.
Vasan K, Werner M, Chandel NS. Mitochondrial Metabolism as a Target for Cancer Therapy. Cell Metab. 2020;32:341–52.
Harrington JS, Ryter SW, Plataki M, Price DR, Choi AMK. Mitochondria in health, disease, and aging. Physiol Rev. 2023;103:2349–422.
Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature. 2021;593:586–90.
Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. 2021;220:e202105043.
Fontana F, Limonta P. The multifaceted roles of mitochondria at the crossroads of cell life and death in cancer. Free Radic Biol Med. 2021;176:203–21.
Zheng S, Wang X, Zhao D, Liu H, Hu Y. Calcium homeostasis and cancer: insights from endoplasmic reticulum-centered organelle communications. Trends Cell Biol. 2023;33:312–23.
He Q, Qu M, Shen T, Su J, Xu Y, Xu C, et al. Control of mitochondria-associated endoplasmic reticulum membranes by protein S-palmitoylation: Novel therapeutic targets for neurodegenerative diseases. Ageing Res Rev. 2023;87:101920.
Booth DM, Enyedi B, Geiszt M, Várnai P, Hajnóczky G. Redox Nanodomains Are Induced by and Control Calcium Signaling at the ER-Mitochondrial Interface. Mol cell. 2016;63:240–8.
von Krusenstiern AN, Robson RN, Qian N, Qiu B, Hu F, Reznik E, et al. Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol. 2023;19:719–30.
Taskén K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. 2004;84:137–67.
Taylor SS, Ilouz R, Zhang P, Kornev AP. Assembly of allosteric macromolecular switches: lessons from PKA. Nat Rev Mol Cell Biol. 2012;13:646–58.
Ramms DJ, Raimondi F, Arang N, Herberg FW, Taylor SS, Gutkind JS. Gαs-Protein Kinase A (PKA) Pathway Signalopathies: The Emerging Genetic Landscape and Therapeutic Potential of Human Diseases Driven by Aberrant Gαs-PKA Signaling. Pharmacol Rev. 2021;73:155–97.
Kritzer MD, Li J, Dodge-Kafka K, Kapiloff MS. AKAPs: the architectural underpinnings of local cAMP signaling. J Mol Cell Cardiol. 2012;52:351–8.
Aggarwal S, Gabrovsek L, Langeberg LK, Golkowski M, Ong SE, Smith FD, et al. Depletion of dAKAP1-protein kinase A signaling islands from the outer mitochondrial membrane alters breast cancer cell metabolism and motility. J Biol Chem. 2019;294:3152–68.
Merrill RA, Strack S. Mitochondria: a kinase anchoring protein 1, a signaling platform for mitochondrial form and function. Int J Biochem Cell Biol. 2014;48:92–6.
Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, et al. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011;44:532–44.
Pryde KR, Smith HL, Chau KY, Schapira AH. PINK1 disables the anti-fission machinery to segregate damaged mitochondria for mitophagy. J Cell Biol. 2016;213:163–71.
Dagda RK, Gusdon AM, Pien I, Strack S, Green S, Li C, et al. Mitochondrially localized PKA reverses mitochondrial pathology and dysfunction in a cellular model of Parkinson’s disease. Cell Death Differ. 2011;18:1914–23.
Yang HM, Kim J, Shin D, Kim JY, You J, Lee HC, et al. Resistin impairs mitochondrial homeostasis via cyclase-associated protein 1-mediated fission, leading to obesity-induced metabolic diseases. Metab: Clin Exp. 2023;138:155343.
Lee AS. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer. 2014;14:263–76.
Maio N, Rouault TA. Outlining the Complex Pathway of Mammalian Fe-S Cluster Biogenesis. Trends Biochem Sci. 2020;45:411–26.
Dai Y, Li F, Jiao Y, Wang G, Zhan T, Xia Y, et al. Mortalin/glucose-regulated protein 75 promotes the cisplatin-resistance of gastric cancer via regulating anti-oxidation/apoptosis and metabolic reprogramming. Cell Death Discov. 2021;7:140.
Zhao Q, Luo T, Gao F, Fu Y, Li B, Shao X, et al. GRP75 Regulates Mitochondrial-Supercomplex Turnover to Modulate Insulin Sensitivity. Diabetes. 2022;71:233–48.
Hayashi T, Rizzuto R, Hajnoczky G, Su TP. MAM: more than just a housekeeper. Trends cell Biol. 2009;19:81–8.
Thoudam T, Chanda D, Lee JY, Jung MK, Sinam IS, Kim BG, et al. Enhanced Ca(2+)-channeling complex formation at the ER-mitochondria interface underlies the pathogenesis of alcohol-associated liver disease. Nat Commun. 2023;14:1703.
Feng S, Huang Q, Deng J, Jia W, Gong J, Xie D, et al. DAB2IP suppresses tumor malignancy by inhibiting GRP75-driven p53 ubiquitination in colon cancer. Cancer Lett. 2022;532:215588.
Liao Y, Liu Y, Shao Z, Xia X, Deng Y, Cai J, et al. A new role of GRP75-USP1-SIX1 protein complex in driving prostate cancer progression and castration resistance. Oncogene. 2021;40:4291–306.
Liu X, Lin W, Shi X, Davies RG, Wagstaff KM, Tao T, et al. PKA-site phosphorylation of importin13 regulates its subcellular localization and nuclear transport function. Biochemical J. 2018;475:2699–712.
Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatol (Baltim, Md). 2016;63:173–84.
Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 2019;23:101107.
Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, et al. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol cell. 2009;34:663–73.
Zhang JZ, Lu TW, Stolerman LM, Tenner B, Yang JR, Zhang JF, et al. Phase Separation of a PKA Regulatory Subunit Controls cAMP Compartmentation and Oncogenic Signaling. Cell. 2020;182:1531–44.e1515.
Haushalter KJ, Schilling JM, Song Y, Sastri M, Perkins GA, Strack S, et al. Cardiac ischemia-reperfusion injury induces ROS-dependent loss of PKA regulatory subunit RIα. Am J Physiol Heart Circulatory Physiol. 2019;317:H1231–h1242.
Carroll WL, Evensen NA. Targeting a major hub of cell fate decisions - the mitochondrial-associated membrane. Haematologica. 2019;104:419–21.
D’Eletto M, Rossin F, Occhigrossi L, Farrace MG, Faccenda D, Desai R, et al. Transglutaminase Type 2 Regulates ER-Mitochondria Contact Sites by Interacting with GRP75. Cell Rep. 2018;25:3573–81.e3574.
Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc Natl Acad Sci USA. 2019;116:25322–8.
Zhang Z, Zhou H, Gu W, Wei Y, Mou S, Wang Y, et al. CGI1746 targets σ(1)R to modulate ferroptosis through mitochondria-associated membranes. Nat Chem. Biol. 2024;20:699–709.
Li Y, Cao Y, Xiao J, Shang J, Tan Q, Ping F, et al. Inhibitor of apoptosis-stimulating protein of p53 inhibits ferroptosis and alleviates intestinal ischemia/reperfusion-induced acute lung injury. Cell Death Differ. 2020;27:2635–50.
Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, Zhuang L, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22:225–34.
Yi J, Zhu J, Wu J, Thompson CB, Jiang X. Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci USA. 2020;117:31189–97.
Wadhwa R, Colgin L, Yaguchi T, Taira K, Reddel RR, Kaul SC. Rhodacyanine dye MKT-077 inhibits in vitro telomerase assay but has no detectable effects on telomerase activity in vivo. Cancer Res. 2002;62:4434–8.
Propper DJ, Braybrooke JP, Taylor DJ, Lodi R, Styles P, Cramer JA, et al. Phase I trial of the selective mitochondrial toxin MKT077 in chemo-resistant solid tumours. Ann Oncol : Off J Eur Soc Med Oncol. 1999;10:923–7.
Britten CD, Rowinsky EK, Baker SD, Weiss GR, Smith L, Stephenson J, et al. A phase I and pharmacokinetic study of the mitochondrial-specific rhodacyanine dye analog MKT 077. Clin Cancer Res. 2000;6:42–9.
Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the Hallmarks of Cancer. Cancer cell. 2018;34:21–43.
Liu H, Zhao D, Li H, Zhang W, Lin Q, Wang X, et al. Blocking iASPP/Nrf2/M-CSF axis improves anti-cancer effect of chemotherapy-induced senescence by attenuating M2 polarization. Cell death Dis. 2022;13:166.
Anandhan A, Dodson M, Shakya A, Chen J, Liu P, Wei Y, et al. NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv. 2023;9:eade9585.
Suzuki T, Yamamoto M. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med. 2015;88:93–100.
Lignitto L, LeBoeuf SE, Homer H, Jiang S, Askenazi M, Karakousi TR, et al. Nrf2 Activation Promotes Lung Cancer Metastasis by Inhibiting the Degradation of Bach1. Cell. 2019;178:316–29.e318.
Wieckowski MR, Giorgi C, Lebiedzinska M, Duszynski J, Pinton P. Isolation of mitochondria-associated membranes and mitochondria from animal tissues and cells. Nat Protoc. 2009;4:1582–90.
Zheng S, Wang X, Liu H, Zhao D, Lin Q, Jiang Q, et al. iASPP suppression mediates terminal UPR and improves BRAF-inhibitor sensitivity of colon cancers. Cell Death Differ. 2023;30:327–40.
Afshar N, Black BE, Paschal BM. Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol Cell Biol. 2005;25:8844–53.
Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 2008;26:1367–72.
Zhao W, Zhao S, Li L, Huang X, Xing S, Zhang Y, et al. Sparse deconvolution improves the resolution of live-cell super-resolution fluorescence microscopy. Nat Biotechnol. 2022;40:606–17.
Acknowledgements
The work was funded by National Nature Science Foundation (No. 82025027, 82150115, 32000517 and 82303023), the National Key R & D Program of China (2022YFA1105200), China Postdoctoral Science Foundation (No. 2022TQ0093, 2020M680045, 2021T140161 and 2023M730871), the Nature Science Foundation of Heilongjiang Province (No. LH2023C070 and YQ2021C024), the Postdoc Foundation of Heilongjiang Province (No. LBH-Z22176), and the Open fund for Key Laboratory of Science and Engineering for the Multi-modal Prevention and Control of Major Chronic Diseases (No. NCD-2023-1-04), Henan Province science and technology research and development plan joint fund (NO. 225200810019).
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The study for human colon cancer samples has been approved by the Research Ethics Committee of Harbin Medical University, China. All animal procedures were performed according to protocols approved by the Rules for Animal Experiments published by the Chinese Government (Beijing, China) and approved by the Research Ethics Committee of Harbin Institute of Technology, China.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Liu, H., Zheng, S., Hou, G. et al. AKAP1/PKA-mediated GRP75 phosphorylation at mitochondria-associated endoplasmic reticulum membranes protects cancer cells against ferroptosis. Cell Death Differ (2024). https://doi.org/10.1038/s41418-024-01414-2IF: 13.7 Q1