
抽象
人体微生物组影响着各种常用处方药的疗效和安全性。设计结合微生物代谢的精准医学方法将需要菌株和分子解析的可扩展计算建模。在这里,我们扩展了我们之前对人类肠道微生物的基因组规模代谢重建的资源,并大大扩展了版本。AGORA2(通过重建和分析组装肠道生物体,第 2 版)涉及 7,302 种菌株,包括 98 种药物的菌株解析药物降解和生物转化能力,并根据比较基因组学和文献搜索进行了广泛策划。微生物重建在三个独立组装的实验数据集上表现非常好,准确度为0.72至0.84,超过了其他重建资源,并以0.81的准确度预测了已知的微生物药物转化。我们证明 AGORA2 通过预测 616 名结直肠癌患者和对照组的肠道微生物组的药物转化潜力来实现个性化、菌株分辨建模,这些患者因个体而异,并与年龄、性别、体重指数和疾病阶段相关。AGORA2 是人类微生物组的知识库,为宿主-微生物组代谢相互作用的个性化预测分析铺平了道路。
其他人正在查看的类似内容

主要
数以万亿计的微生物栖息在人体胃肠道中,个体间差异很大,具体取决于性别、年龄、种族、生活方式和健康状况等因素1.肠道微生物群合成生物活性代谢物,如短链脂肪酸、激素和神经递质2,并参与常用处方药的代谢3,导致药物失活、活化、解毒或再毒化4.人类肠道微生物已被证明可以代谢 271 种测试药物中的 176 种5,活动因人而异6.因此,人们提出了将饮食、遗传学和微生物组考虑在内的精准医学干预措施7.预测这种个性化治疗需要详细了解药物转化反应在人类微生物分类群中的分布以及这些转化的化学计量。
一种包括代谢的详细化学计量表示的机械系统生物学方法是基于约束的重建和分析 (COBRA)8.COBRA依赖于目标生物体的基因组规模重建,这些重建通常是根据现有文献手动策划的8.通过应用特定条件的约束,这些重建可以转换为预测计算模型9,包括(元)组学和营养数据,并链接在一起以询问菌株解析的个性化微生物组模型10,11.因此,COBRA方法非常适合探索代谢的人类微生物组彗代谢12,13.促进人类中数千种已知物种的基因组规模重建14、半自动重建工具,如 CarveMe15、MetaGEM16, 米格雷内17和 gapseq18,已出版。尽管它们具有许多优点,但这些工具对手动改进的基因组注释和来自同行评审文献的实验数据的策展提供了有限的支持。两者对于纳入尚未常规注释的物种特异性途径(例如,药物代谢)都至关重要9.为了克服这些局限性,我们开发了一种半自动策展流程,以手动组装的比较基因组分析和实验数据为指导19,这之前使AGORA的生成成为可能,AGORA是人类肠道微生物菌株的773个基因组规模重建的资源,代表605个物种和14个门20.
在这里,我们介绍了AGORA的范围和覆盖范围的扩展,称为AGORA2,包括7,302个菌株,1,738个物种和25个门的微生物重建。AGORA2总结了通过人工比较基因组学分析以及文献和教科书审查获得的知识和实验数据,并针对三个独立收集的实验数据集表现出高精度。AGORA2 已通过手动配制的分子和菌株分离药物生物转化和降解反应进行扩展,涵盖 5,000 多种菌株、98 种药物和 15 种酶,其中一些已根据独立实验数据进行了验证。AGORA2 重建与通用21以及器官分辨的、性别特异性的、全身的人类代谢重建22.我们演示了使用 AGORA2 预测 616 名个体的个性化肠道微生物药物代谢。综上所述,AGORA2 重建可以单独使用或一起用于研究计算机中的微生物代谢和宿主-微生物群彗出代谢。
结果
数据驱动的各种人类微生物重建
为了重建 AGORA2 纲要(补充表 1)中的 7,302 种肠道微生物菌株,我们大幅修订和扩展了(方法)先前开发的20数据驱动的重建细化管道,被视为 DEMETER(Data-drivEn METabolic nEtwork Refinement)19.总体而言,DEMETER工作流程包括数据收集、数据集成、重建草稿生成以及将反应和代谢物转化为虚拟代谢人(VMH)23命名空间,并同时进行迭代细化、填空和调试19.重建细化遵循标准操作程序,以生成高质量的重建9并通过测试套件不断验证19(附表2和附注2)。
扩大分类覆盖范围后(图1)。1a,b,补充表1和补充说明1)并检索相应的基因组序列,我们通过在线平台KBase生成了自动草图重建24,随后通过DEMETER管道进行了改进和扩展19 (方法)。由于缺乏准确的基因组注释是基因组规模重建预测潜力不确定性的来源25,我们使用 PubSEED 手动验证并改进了 7,302 个 (74%) 基因组中 5,438 个代谢子系统中 446 个基因功能的注释26(补充表3a-d)。为了进一步确保物种特异性代谢能力的准确表示,我们进行了广泛的手动文献检索,涵盖 732 篇同行评审论文和两本微生物参考教科书,获得了 7,302 种菌株中的 6,971 种 (95%) 的信息(方法)。对于剩余的331株菌株,要么没有实验数据,要么文献中报道的所有生化测试均为阴性。在收集数据的驱动下进行的广泛改进导致每次重建平均增加 685.72 个(标准差:±620.83)个反应并去除 685.72 个(标准差:±620.83)个反应(补充图 1)。1).重建草案中提供的生物质反应被整理,并在适当的情况下将反应放置在周质隔室中(补充说明3)。此外,我们检索了 3,613 种代谢物中 1,838 种 (51%) 代谢物的代谢结构,并为 AGORA2 捕获的 8,637 种 (65%) 酶促和转运反应中的 5,583 种提供了原子-原子图谱(方法)。由于这些广泛的策展工作,从精细重建中得出的代谢模型显示出其预测潜力明显优于从KBase草案重建中得出的模型(图1)。1c,d和补充说明2)。作为对重建质量的额外评估,我们为所有重建(方法)生成了一份公正的质量控制报告,平均得分为 73%。

a, 生物分类覆盖率和重建菌株的来源。 b, 7,302株菌株的分类分布。 c, AGORA2 重建和 KBase 草图重建的特征。c: 胞质溶胶;e: 细胞外空间;p, 周质。西方饮食 (WD) 和无限培养基 (UM) 的生长率以 h 为单位−1(方法)。WD 的 ATP 生产潜力以每克 mmol 为单位干重每小时。显示的是所有模型的平均值±标准差。d,从比较基因组学和文献中获得积极发现的重建数量,以及与相应生物体的发现一致的策划和重建草案的百分比。N/A,不适用,因为重建草案中没有该途径。CM,化学成分确定的培养基。
然后,我们按分类分布对AGORA2重建的内容进行聚类。总体而言,AGORA2反映了捕获菌株的多样性,因为它们根据其反应覆盖率按类别和家族聚类(图1)。图2a,b,补充图。3a和补充说明4)。芽孢杆菌属和伽马变形菌属中的几个属形成了亚群,说明了它们之间的重要代谢差异(图1)。2c,d, 补充图。2a、b和补充说明4;Kruskal-Wallis 检验:P = 0.0001)。跨门代谢差异也转化为重建大小和预测生长速率的差异(图1)。2e-h)以及它们消耗和分泌代谢物的潜力(补充图。3a,b)。综上所述,源自AGORA2的模型捕获了重建微生物的分类群特异性代谢特征。

a–d, 通过t-SNE聚类52每次重建的所有途径中的反应存在。不同分类单位的坐标在统计学上存在差异(Kruskal-Wallis检验,P = 0.0001)。a, 最大类的成员。b, 最大家庭的成员。c, 按属划分的杆菌类成员。d, 按属划分的 Gammaproteobacteria 类的成员。e–h,跨门所有AGORA2重建的特征:e,反应数。f, 代谢物数。g, 基因数。h, 生长率(h)−1有氧西式饮食。
AGORA2 可针对三个独立的数据集进行预测
虽然可以快速生成自动草稿重建,但它们仍然需要后续的策展工作才能具有预测性27.几种(半)自动重建工具弥合了自动绘制和完全手动策划的重建之间的差距,包括 CarveMe15、加普塞克18和 MIGRENE17.为了进一步了解 AGORA2 和 DEMETER 管道的质量,我们将 AGORA2 的预测潜力和模型特性与其他微生物基因组规模重建资源进行了比较。为此,我们检索了 8,075 个通过 gapseq 构建的重建18,通过MIGRINE建造的1,333个重建,被视为MAGMA17,以及存放在 BiGG 数据库中的 72 个手动策划的基因组规模重建28.此外,我们还构建了 CarveMe157,279 株 AGORA2 菌株和 gapseq 的重建181,767 个 AGORA2 菌株子集的重建(方法)。
为了对重建质量进行公正的评估,我们首先确定了通量一致反应的分数29在每个资源中。只有来自BiGG的手动重建和通过CarveMe构建的重建具有比AGORA2更高的通量一致性反应比例(图1)。3a,b;第 < 1 × 10−30,Wilcoxon 秩和检验)。请注意,我们的重建代表知识库;因此,如果存在基因或反应的遗传或生化证据,它将被包括在重建中。相比之下,CarveMe的设计消除了代谢重建中所有通量不一致的反应15.与KBase草图重建相比,尽管AGORA2的代谢含量较大,但其通量一致性反应的百分比显著高于gapseq和MAGMA(图1)。3a,c;第 < 1 × 10−30,Wilcoxon 秩和检验)。还观察到,除AGORA2和gapseq外,所有资源都产生了非常高的ATP(高达1,000 mmol g干重−1h−1)在复杂介质上至少用于模型子集(图1)。3b,c)。因此,在这些模型中,ATP生产通量仅受反应上限的限制,这通常表明存在徒劳循环9.

比较了 7,302 个 AGORA2 和 KBase 重建草案,72 个来自 BiGG 数据库的手动策划重建28,通过 CarveMe 重建了 5,587 个重建项目15,通过 gapseq 重建了 8,075 个重建项目18和 1,333 次岩浆重建17.a,在化学计量和通量上一致的反应分数,如参考文献中定义。29对于从五个比较资源派生的每个模型。根据定义,在化学计量上不一致的交换和需求反应被排除在外。b,在复杂培养基上产生好氧和厌氧ATP(mmol/g干重per h) 由从五个比较资源中得出的每个模型。c,比较资源的重建属性概述。d,针对三个独立的实验数据集验证AGORA2、KBase、BiGG、CarveMe、gapseq和MAGMA的模型数量和预测数量概述30,32,33.e, 在三个实验数据集中,五种资源在预测摄取和分泌方面的总体精度具有 95% 置信区间的条形图。使用代谢模型作为随机效应变量,通过混合效应logistic回归确定预测准确性的显著性,以解释来自同一模型的预测的统计依赖性。NA 表示由于空类别(例如,未检测到真负值)而缺少 P 值。f, 三个实验数据集上各种资源的每种模型的精度比较。P值由符号秩检验得出。
基因组规模重建最关键的方面是其捕获目标生物体已知生化或生理特征的准确性9,也就是说,它有可能做出生物学上合理的预测。因此,我们着手确定AGORA2的预测潜力。为了进行无偏倚的评估,我们从三个不同的来源(方法)检索了生物体特异性的实验数据。首先,我们从 NJC19 资源中检索了 AGORA2 中 455 个物种(5,319 个菌株)的物种水平正负代谢物摄取和分泌数据30.请注意,NJC19的前体NJS16(参考文献。31),仅包含阳性数据,已用于提炼AGORA2。接下来,我们绘制了从Madin等人那里检索到的物种水平的正代谢物摄取数据。32,适用于 AGORA2 中的 185 个物种(328 个菌株)(“Madin”数据)。最后,我们从 BacDive 数据库中检索了 676 株 AGORA2 菌株的菌株分辨阳性和阴性代谢物摄取和分泌数据,以及 881 株 AGORA2 菌株的阳性和阴性酶活性数据33.在 AGORA2 的改进过程中,Madin 数据集和 BacDive 都没有使用。对于代谢物的摄取和分泌,AGORA2重建很好地捕获了目标生物体的已知能力(对NJC19、BacDive和Madin的总体准确度分别为0.82、0.81和0.84;无花果。3e和附表4d)。对于酶活性,精度略低,为0.72(图1)。3e和附表4d)。AGORA2 的特异性低于 NJC19 上的其他资源。然而,在 AGORA2 中观察到的大多数假阳性与大肠杆菌中的谷氨酸摄取有关(补充表 4c),这是 NJC19 数据集中基于单个大肠杆菌菌株报告的阴性发现。
然后,在可能的情况下,我们将AGORA2与其他四种资源的预测潜力进行了比较。在 7,302 个重建的 AGORA2 菌株中,7,279 个是通过 CarveMe 重建的,451 个与通过 gapseq 构建的重建重叠,60 个与 BiGG 数据库中可用的重建菌株重叠(补充表 4a)。没有菌株与MAGMA重叠,因为它由宏基因组组装的基因组构建的泛物种重建组成17,但可以在物种水平上绘制216个重建图(补充表4a)。对于四个资源和每个数据集,我们随后计算了与AGORA2重叠的生物体的预测潜力(图1)。3d-f和补充表4b-d)。虽然 MAGMA 和 AGORA2 在 NJC19 和 BacDive 数据集上实现了显著的分泌和摄取预测精度,但 KBase 在 NJC19 中未能在代谢物摄取和分泌方面表现得优于机会,而 CarveMe 未能在 NJC19 数据集中预测显着分泌(图 1)。3e和附表4d)。本研究中针对 AGORA2 菌株子集构建的 gaqseq 重建与作者发表的一组 gapseq 重建相当18(补充表4b)。
为了直接比较AGORA2与KBase、CarveMe、gapseq、BiGG和MAGMA的性能,我们分别计算了每个模型的摄取和分泌精度。然后,我们通过非参数符号秩检验比较了 AGORA2 和每个资源重叠的模型的精度。在所有三个数据集上,AGORA2 都明显优于所有其他方法,除了 BacDive 数据上的 BiGG 模型重叠太小而无法获得足够的统计功效,以及 BacDive 酶数据上的 gapseq,其性能与 AGORA2 相当(71% 对 72%;无花果。3e,f)。
总而言之,AGORA2重建很好地捕获了各自生物体的已知特征,超过了其他半自动生成的重建,并且可与手动策划的重建相媲美。这些结果证明了在AGORA2开发过程中,在物种-物种实验数据的指导下进行的广泛策展工作改进的价值,如上所述。因此,AGORA2在代谢物摄取和分泌数据方面表现特别好,这些数据需要基于实验数据进行整理,而酶活性数据可以基于基因组注释进行整理。剩余的假阳性和假阴性预测(补充表4c)将在未来的工作中按照迭代策展理念加以解决9.通量不一致的反应,表明它们含有死端代谢物29,可以作为填补空白工作的起点,从而实现生物发现34.
基因组和文献组引导的微生物药物代谢
微生物可以通过降解(例如水解)和生物转化(例如还原)直接或间接影响药物活性和毒性3,4.然而,药物代谢仅通过基因组注释管道在有限程度上捕获,并且以前没有对药物代谢酶进行系统的比较基因组分析。因此,微生物药物转化尚未被任何现有的基因组规模重建资源捕获。为了填补这一空白,我们对 25 个药物基因进行了广泛的手动比较基因组分析,编码了 15 种被证明直接或间接影响药物代谢的酶(补充表 5a)、它们的亚细胞位置和 12 个编码药物转运蛋白的基因(补充表 3b)。所有 5,438 个分析菌株都携带至少一种药物代谢酶(补充表 3c)。由于这些酶也参与中枢代谢,例如核苷代谢,因此这种高覆盖率是预期的。然后,我们对属于10个药物组和32个亚组的98种常用处方药的代谢物结构、配方和费用进行了彻底的文献和数据库回顾(补充表5b)。我们配制了含有363种代谢物的1,440种药物相关反应(补充表6a,b),并根据基因组证据平均在重建中添加了188种药物相关反应和111种代谢物。我们验证了准确度为 0.81(灵敏度:0.87,特异性:0.74,Fisher 精确检验:P = 2.01 × 10−23,混合效应逻辑回归考虑了来自同一模型的预测的随机依赖性:P = 1.209 × 10−07),根据独立发表的253对药物-微生物的实验数据进行药物代谢预测(补充表7和图1)。18个假阳性预测可能表明非功能性基因或调控机制,而31个假阴性预测可能是由于基因组的不完整或完整基因组中的非直系同源位移,或者目前未知的编码反应的同源物。

药物代谢能力的分类分布
接下来,我们分析了注释的药物和转运基因的分类分布(图1)。4b-d和补充表3c)。在编码参与药物代谢的基因的 14 个分析门中,每个门中至少有一个菌株(图 1)。最普遍的药物代谢酶是胞苷脱氨酶和硝基还原酶,分别存在于12门和13门(补充图13)。7a,b)。另一种中枢代谢酶,嘧啶核苷磷酸化酶,也广泛分布,但单系分支特异于brivudine和sorivudine的代谢35仅在拟杆菌门中发现(图1)。图4c,d和补充图。许多药物通过添加葡萄糖醛酸由肝脏解毒,这种修饰被微生物β-葡萄糖醛酸酶逆转4.该酶存在于>99%的分析大肠杆菌菌株中,并且还广泛分布在拟杆菌门和厚壁菌门菌株中(图1)。图4c,d和补充图。7d),与先前的分析一致36.大肠杆菌是药物代谢最富集的物种,>99%的分析菌株携带7至10种药物酶(补充表3c)。总而言之,药物代谢酶和转运蛋白分布广泛,但存在重要的门特异性和菌株特异性差异。为了阐明这些药物代谢能力可能赋予微生物的潜在益处,我们计算了药物降解的菌株特异性能量、碳和氮产量。该分析表明,分布在门中的许多菌株能够使用药物作为能量、碳和/或氮的来源(补充图)。4和补充表8)。
药物代谢能力的个性化建模
由于人类微生物不是孤立存在的,我们解决了一个重要的问题,即个体肠道微生物组之间的总药物代谢能力有何不同。先前开发的社区建模框架10允许对整个社区的代谢能力以及生物体对粪便代谢物水平的贡献进行可扩展、可处理的计算37.我们使用了来自日本365名结直肠癌(CRC)患者和251名健康对照者的宏基因组数据集38这使我们能够询问每个肠道微生物组的代谢能力,并根据代谢组学数据验证通量37.总共有97%的命名物种可以映射到AGORA2上(而AGORA为72%)。对于每个人的肠道微生物组,我们建立并询问了一个社区模型(方法),从而预测了总药物代谢潜力(图1)。5a和补充表9)。对于某些酶,例如二氢嘧啶脱氢酶和多巴胺脱羟化酶,药物转化电位仅与相应药物代谢反应的总丰度表现出有限的相关性,表明存在通量限制代谢瓶颈(图1)。分析这些瓶颈需要模拟其代谢环境中的酶功能。影子价格分析(方法)显示,在左旋多巴降解为间酪胺等两步反应中,第二步的药物转化潜力受到执行第一步的物种丰度的限制(补充说明5,补充图5)。6和补充表10)。已知左旋多巴降解是由不同物种进行的两步途径39(补充图。6).

a,365 名日本 CRC 患者和 251 名日本平均饮食对照组微生物组中的药物转化潜力。小提琴图显示了药物代谢物通量的分布,单位为每人每 d. b,药物转化潜力(每人每 d)与 616 个微生物组中产生所示药物代谢物的反应的总相对丰度作图。有关每种药物代谢酶的描述,请参阅补充表5a。
虽然大多数药物可以被至少95%的微生物组在计算机中定性代谢,但只有53%的微生物组具有代谢地高辛的能力,而左旋多巴可以被86%的研究微生物组代谢成多巴胺,46%被代谢成间酪胺(图1)。地高辛转化和左旋多巴降解的第二步都严格依赖于Eggerthella lenta的存在(补充图1)。8),并且已知会降低药物的生物利用度4,39.此外,虽然除三种微生物组外,所有微生物组都可以通过偶氮还原酶活性激活抗炎性肠病(IBD)前药巴柳氮,但任何微生物组达到的活性形式的巴柳氮(5-氨基水杨酸)的最高分泌通量为339.81 mmol d−1每人,而平均值为 25.47 ± 40.84 mmol d−1每人(图1)。这种变异可能具有很高的临床相关性,因为它表明并非所有微生物组都能同样激活巴柳氮。作为敏感性分析,我们使用平均欧洲饮食而不是日本饮食重新计算了药物代谢能力,发现所有药物的药物代谢能力几乎没有改变,因此对饮食限制具有高度鲁棒性(补充图)。7).
微生物组水平的通量对临床参数敏感
接下来,我们研究了药物代谢能力是否与CRC相关。对于包括抗癌药物在内的所有药物,尽管在CRC宏基因组中报告了29个物种的富集,但在经过多次测试校正后,药物代谢能力均未发现定性和定量差异40.在标称水平(P < 0.05)上,亚硝基氯霉素在癌症病例中增加(图1)。尽管如此,由于微生物群组成不同,无论疾病状态如何,药物代谢潜力都存在巨大的个体差异(图6a)。5a)。

a,模拟药物代谢物的描述性统计概述。b,各种药物代谢物随年龄变化的散点图(红色,对照;蓝色,癌症),病例和对照组为非线性回归线。回归线是用受限的三次样条估计的。所有回归模型的P<为0.0001(FDR<为0.05),病例和对照组的回归系数几乎相同。c, L-乳酸、L-蛋氨酸和γ-氨基丁酸的粪便种类代谢物标志预测。上图表示微生物群落净分泌通量的计算机变化散点图,这些变化来自群落建模,与依赖于微生物物种存在的测量粪便浓度的变化。每个点代表一种对代谢物浓度有影响的微生物物种,P至少<0.05。下图描绘了通过计算机模拟进行符号预测的混淆矩阵。由于物种-物种和代谢物-代谢物的相互依赖性,应谨慎对待从 Fisher 精确检验得出的 P 值。
最后,我们研究了年龄、性别和体重指数(BMI)与微生物组药物代谢能力的统计关联模式(图1)。图6b和补充图6b。9). 药物代谢物的五种预测分泌潜力与年龄明显相关(图1)。6b),尽管效应量为小到中等(解释方差<8%)。例如,索利夫定转化为有毒副产物与年龄呈非线性关联,分泌能力从60岁开始下降(图1)。6b;R型2= 0.047,P = 7.17 × 10 −06).女性牛磺胆酸盐的代谢能力显著提高,化疗药物吉西他滨的转化潜力略有降低,但显著降低(补充图1)。总之,我们的分析能够研究与肠道微生物组的药物代谢能力相关的临床参数。
群落模型预测物种-代谢物关联
作为验证的最后一步,我们测试了基于 AGORA2 的群落建模是否能够预测 CRC 样品中微生物物种存在与粪便代谢物浓度之间的统计关联迹象,遵循之前建立的程序41.我们计算了 52 种 AGORA 代谢物(方法)的粪便净分泌率,这些代谢物的粪便代谢组学数据来自同一日本队列38.由于这些代谢组学数据未用于构建基于AGORA2的群落模型,因此该过程代表了独立的验证。
经过多次测试校正后,基于AGORA2的群落建模可预测52种代谢物中24种代谢物中显著的物种-代谢物关联迹象(图1)。6c和补充表11),P<为0.05,19为错误发现率(FDR)<0.05。氨基酸和已知的发酵产物(例如,l-乳酸盐、丁酸盐)以及胺类物质(补充表11)被特别充分地覆盖。值得注意的是,对于某些代谢物,例如蛋氨酸(图1)。6c),体内关联统计量始终与相应的计算机结合统计量相反。后一种结果可能对应于微生物群落对代谢物的净摄取。非显著性符号预测,如图所示。γ-氨基丁酸盐的 6c 可能有多种原因,从主导粪便浓度变化的宿主因素到不完整的群落模型或统计模型中缺少混杂因素,导致体内关联假阳性。综上所述,基于AGORA2的群落模型可以预测多种代谢物的物种代谢物关联方向,突出了模型的预测性质。
讨论
在这里,我们介绍了 AGORA2,这是一个包含 7,302 个人类相关微生物基因组规模重建的资源,据我们所知,其覆盖范围、范围和管理工作是前所未有的。AGORA2遵循系统生物学研究界制定的质量标准9,42,准确捕获目标生物的生化和生理特征,超越其他重建资源,并包括手动精炼的菌株分辨药物代谢能力。它可以通过专用的计算管道对人类微生物代谢进行个性化建模10,最近在计算效率和实现功能方面得到了改进43.因此,使用AGORA2的个性化微生物组建模可以在合理的时间范围内在标准个人计算机上进行(方法)。
微生物联盟的计算建模越来越被认为是体外和体内实验的补充方法,并且可以产生可实验检验的假设13,44.我们对肠道微生物的了解仍然有限,因此,任何计算机重建本质上都是不完整的,需要定期更新45.例如,最近的一项研究发现,在271种测试药物中,有176种可以被人类细菌代谢,并且对于这些药物的一部分,转化可能与特定的基因功能有关5.通过未来的比较基因组学以及代谢物和反应制剂工作,AGORA2可能会通过这些药物转化进行扩展,以进一步扩大其对处方药代谢的覆盖。由于 AGORA2 使用相同的代谢物和反应命名法23作为人体代谢重建21以及全身代谢重建22,它可用于预测整体宿主-微生物组代谢及其对人体器官水平代谢的潜在贡献22.
迄今为止,AGORA已经完成了近50项模拟微生物-微生物、宿主-微生物和微生物组相互作用的研究46,以及与可用的软件工具一起10,47,为基于约束的多物种相互作用建模的规模和范围的最新进展做出了重大贡献46.然而,AGORA在一定程度上受到其有限的分类覆盖范围的阻碍,其中主要包括西化的肠道微生物组20.相比之下,AGORA2 还捕获非西化微生物组以及皮肤、口腔和阴道微生物组中常见的微生物;包括许多未培养的微生物;并且与宏基因组组装基因组的几种资源中报告的物种高度重叠(补充说明1)。总之,这种扩展提高了微生物组水平模型的预测保真度,包括非肠道和非西化微生物组。
我们报告了CRC患者特异性微生物药物转化能力与临床参数(如年龄和BMI)之间的关联(图1)。巴柳苷是一种用于治疗IBD的抗炎药,它展示了AGORA2如何用于为临床研究提供信息,并可能促进治疗的个性化。巴柳氮在溃疡性结肠炎中具有诱导缓解 (NNT: 10) 和维持 (NNT: 6) 的需要治疗 (NNT) 指标48,表明大多数患者不会从该药物中获利。一致地,巴柳苷的活化电位在所研究的CRC队列微生物组中变化很大(图1)。5a),表明并非所有人都能从巴柳嗪治疗中平等获益。因此,我们建议AGORA2与宏基因组学相结合可以预测IBD患者分为巴柳嗪反应者和无反应者,然后可以在后续临床试验中得到验证。药物代谢能力与年龄组、BMI和性别相关的发现(图1)。图6b和补充图6b。9) 表明 AGORA2 与社区建模相结合可用于大型流行病学队列研究,将预测的代谢通量与临床参数联系起来,从而为了解微生物组在改变健康风险和导致不良健康结果方面的作用开辟了新的研究可能性。最后,基于AGORA2的群落模型能够预测一系列代谢物的物种-代谢物关联方向(图1)。6c),证明在提供微生物组代谢性状的有效计算机标记物方面具有实用性。
综上所述,我们提出了基因组规模的代谢重建资源AGORA2,它准确地捕获了生物体的特异性能力,并可用于构建预测性的个性化微生物组模型。AGORA2 以及本研究中使用的所有工具和脚本均免费提供给研究界。我们预计,与其前身AGORA2类似,微生物组和基于约束的建模社区将非常感兴趣,并具有更广泛的潜在应用46.作为一项独特的功能,AGORA2 可捕获菌株分离的微生物药物代谢。预测药物对实际药物浓度的反应将需要混合建模方法,例如,将基于约束的建模与基于生理学的药代动力学建模集成在一起49,50.使用基于约束的器官分辨全身代谢模型与肠道微生物组模型集成22,并使用这种混合建模方法、膳食补充剂、益生菌或微生物组靶向干预措施,这些方法已被证明可以减轻药物的副作用4,可以预测和验证49.因此,AGORA2 为一种集成的、多尺度的建模方法铺平了道路,该方法可以实现计算机临床试验49,51并为精准医疗做出贡献。
方法
新重建生物体的选择和全基因组序列的检索
首先,我们检索了 PubSEED 上可用的 4,185 个人类肠道相关菌株的基因组53(补充说明6)。为了扩大物种覆盖范围,我们对从人类微生物组中分离或检测到的物种进行了广泛的文献检索,并提供了可用的全基因组序列(补充表1)。这次搜索又增加了1,324个菌株,其中包括127个小鼠相关菌株的基因组。从美国国家生物技术信息中心(NCBI)FTP站点(ftp://ftp.ncbi.nlm.nih.gov/)以FASTA格式检索相应的全基因组序列。此外,我们纳入了 26 个 Eggerthella lenta 菌株的基因组54可在 https://www.ncbi.nlm.nih.gov/bioproject/PRJNA412637 购买。最后,我们从人类胃肠道细菌培养物库中检索了 761 个人类微生物基因组55来自 https://www.ebi.ac.uk/ena/data/view/PRJEB23845 和 https://www.ebi.ac.uk/ena/data/view/PRJEB10915 的 FASTQ 格式。与从 VMH 获得的 AGORA1.03 一起23,这些共同努力导致 7,302 个菌株和 1,738 个物种被纳入 AGORA2。
通过比较基因组学手动改进代谢途径和基因注释
在分析的 7,302 种菌株中,PubSEED 资源中存在 5,438 种细菌菌株和 3 种古菌菌株53,56(补充说明6),并可以通过比较基因组学重新注释其代谢功能。总共 34 个代谢亚系统,这些子系统之前已针对较小的肠道微生物菌株子集进行了重建票价:20、57、58、59、60,以及新创建的药物代谢子系统,被考虑用于分析(补充表3a为子系统的完整列表)。所有子系统都可以在 PubSEED 网站上找到。
子系统的管理
为了注释每个子系统中的基因,使用了 PubSEED 平台53.每个子系统的功能角色根据 (1) 蛋白质的规定功能角色,(2) 蛋白质与先前确认的功能角色的蛋白质的序列相似性以及 (3) 基因组背景(补充说明 7)进行注释。
比较基因组学分析的代谢途径注意事项
通路中缺少一种或多种酶的基因可能导致代谢重建中的反应受阻。为了避免这种情况,我们在基因组注释期间估计了代谢途径的完整性。对于每种可能合成的代谢物,所有生物合成途径的收集都与KEGG PATHWAY资源一致61子系统的基因归因于代谢途径的相应步骤。没有随后的反应被确定为间隙。只有间隙不超过两个且间隙长度不超过一个步骤(补充说明8)的途径才被进一步填充并用于生成反应。
基于序列的间隙填充
对于间隙通路,双向最佳命中 (BBH) 方法62使用:(1)与间隙相对应并存在于相关生物(属于同一物种、属或科)基因组中的基因用作在具有间隙的基因组中进行BLAST搜索的查询。(2) 可能的 BBH 被定义为与 e 值 -50 和蛋白质同一性 ≥50% 的查询蛋白比对的同源物<。(3)对于每个可能的BBH,对作为查询蛋白来源的基因组进行反向搜索。(4)如果查询蛋白及其在分析基因组中的最佳同源物形成BBH对,则填补了空白。(5) 查询蛋白及其直系同源物的类似基因组背景被认为是对已鉴定的 BBH 对的正字学的额外确认。
药物代谢基因的注释
为了注释药物代谢基因,我们使用了以下管道。(1)从科学文献中鉴定已知编码一系列微生物中药物代谢酶的基因(补充表5a)。(2)利用这些已知药物代谢基因的氨基酸序列作为查询,对每个分析的基因组进行了BLAST搜索。(3)然后将得到的最佳BLAST命中用作在具有已知药物代谢基因的基因组中进行BLAST搜索的查询,以确认已知的蛋白质序列及其最佳BLAST命中形成一对BBH。 (4)所有BBH都用于构建有根的最大似然树。(5)将所有先前已知的蛋白质定位到树上,并确定所有含有已知药物代谢酶的单系分支(补充图1)。(6)这些分支中所有带注释的蛋白质都被认为是已知药物代谢蛋白的直系同源物。所有与已知药物代谢蛋白不在分支中的蛋白质都被认为是具有其他功能的蛋白质,并被排除在进一步分析之外。随后,再次构建了一棵树,用于已知药物代谢蛋白的直系同源物。(7)对于l-酪氨酸脱羧酶(TdcA,酶委员会(EC)4.1.1.25)和胞苷脱氨酶(cCda,EC 3.5.4.5),我们发现基因组背景在物种之间是保守的,我们还分析了基因组背景。如果候选基因的基因组背景与已知药物代谢基因的基因组背景相似,则该候选基因被认为是已知蛋白质的直系同源物。否则,它被认为是一个假阳性预测,并被排除在进一步的分析之外(补充说明9和补充图9)。10). 至于(6),仅针对已知蛋白质的直系同源物再次构建树。(8)对于每棵树,仅包括已知基因的直系同源物,我们定义了仅包含来自一个物种的蛋白质的单系分支。对于每个这样的物种特异性分支,我们使用 CELLO v.2.5 系统 (cello.life.nctu.edu.tw) 预测了亚细胞定位(补充说明 10)。(9)对于细胞质酶,根据基因组背景预测药物转运蛋白(补充说明11和补充表3b)。
工具
PubSEED 平台53,56用于注释子系统。要搜索先前已知蛋白质的 BBH,BLAST 算法63使用了在 PubSEED 平台中实现的。此外,PubSEED 平台还用于分析基因组背景。为了分析蛋白质结构域结构,我们检索了保守结构域数据库(CDD)64使用以下参数:E 值≤ 0.01,最大命中数等于 500。为了预测蛋白质亚细胞定位,CELLO65使用了网络工具。使用 MUSCLE v.3.8.31 (ref.66).对于每一次多重对齐,都使用 Clustal X 评估位置质量分数67,68.此后,将所有得分为零的位置从比对中移除,并将修改后的比对用于构建系统发育树。系统发育树是使用最大似然法构建的,默认参数在 PhyML-3.0 中实现(参考文献)。69).获得的树是中点根的,并使用交互式查看器 Dendroscope, v.3.2.10, build 19 (ref.70).
文献和数据库检索
通过在 PubMed 中输入 AGORA2 物种的名称来检索生化和生理表征论文 (https://www.ncbi.nlm.nih.gov/pubmed/)。随后,从732篇同行评审论文和>8,000页的微生物参考教科书中手动提取了132种碳源、30种发酵途径、64种生长因子、73种代谢物的消耗和51种代谢物的分泌信息71.此外,通过数据库检索检索了每个重建菌株的分类学、形态学、代谢和基因组大小等性状。菌株的分类学分类取自NCBI分类法(https://www.ncbi.nlm.nih.gov/taxonomy/)。从综合微生物基因组和微生物组中手动检索有关形态、栖息地、身体部位、革兰氏状态、氧状态、代谢、运动和基因组大小的信息72数据库(https://img.jgi.doe.gov/)(补充表1)。所有用于精炼AGORA2的实验数据均可在 https://github.com/opencobra/COBRA.papers/tree/master/2021_demeter/input 获得。
生成重建草稿
通过KBase生成重建草稿24叙述界面。KBase中存在的基因组被直接导入到叙述中。否则,FASTA格式的基因组被上传到暂存区,随后通过“从暂存区批量导入组装”(https://narrative.kbase.us/#catalog/apps/kb_uploadmethods/batch_import_assembly_from_staging)应用程序导入到叙述中。从 https://www.ebi.ac.uk/ena/data/view/PRJEB23845 和 https://www.ebi.ac.uk/ena/data/view/PRJEB10915 检索到相应文件的链接后,通过“从 Web 导入配对端读取”(https://narrative.kbase.us/#catalog/apps/kb_uploadmethods/load_paired_end_reads_from_URL) 应用程序将 FASTQ 格式的基因组直接导入叙述中。导入的程序集使用 RAST 子系统进行注释73通过“注释多个程序集”(https://narrative.kbase.us/#appcatalog/app/RAST_SDK/annotate_contigsets)应用程序。通过“创建多个代谢模型”(https://narrative.kbase.us/#appcatalog/app/fba_tools/build_multiple_metabolic_models)应用程序生成代谢重建草图,并通过“批量下载建模对象”(https://narrative.kbase.us/#appcatalog/app/fba_tools/bulk_download_modeling_objects)应用程序以SBML格式导出。
半自动化、数据驱动的优化管道
我们开发了一种半自动精炼管道 DEMETER19,它以前曾用于构建AGORA20.简而言之,DEMETER是通过在少数重建中测试间隙填充步骤并将已确定的解决方案传播到许多重建中而开发的。在DEMETER中,通过用每个实验证明的功能的完整途径填充适当的重建间隙来对实验数据进行管理。在好氧和厌氧条件下以及在规定的培养基上生产生物质以及细胞壁成分的生物合成也可以通过以前在少数重建中确定的间隙填充溶液来实现。同样,通过在少量重建中识别和纠正受影响的反应并在 DEMETER 的开发过程中传播这些变化来解决徒劳的循环。有关DEMETER的更多详细信息,请参阅参考文献。19.COBRA 工具箱中提供了详细的教程47.
对于AGORA2的生成,我们大幅修改了DEMETER。具体来说,我们 (1) 将 ~1,000 个额外的反应和 ~800 个代谢物从 KBase 翻译成 VMH23命名法;(2)在必要时引入额外的间隙填充反应,以便在缺氧条件下在具有热力学一致反应方向的复杂介质上生产生物质;(3)通过使负责任的反应不可逆,消除了导致热力学上不可信的ATP产生的徒劳循环;(4)通过填充间隙和/或删除适当的反应来确保所有重建都捕获了收集的实验数据;(5)调整生物量目标函数,以解释特定类别的细胞膜和细胞壁结构,并引入周质区室(补充说明3)。如前所述20,所有优化和调试解决方案都是针对重建的子集手动确定的,随后根据需要传播到许多重建中。所有新纳入的代谢物和反应均基于文献和/或数据库配制23,28,74搜索,同时通过重建工具rBioNet确保质量和电荷平衡75.通过比较基因组学(补充表3b,c)鉴定的反应被添加到多达5,438个重建中。非基因相关反应,即通过比较基因组学无法找到相应基因,如果这样做不会消除生物质生产,则从重建草案中删除。
策展工作通过测试套件进行了验证19.具体来说,它系统地测试了每个重建(1)是否在复杂培养基上厌氧生长;(2)具有正确的重建结构,即质量和电荷平衡,以及正确的基因-蛋白质-反应关联语法;(3)在热力学上是可行的,例如,产生实际量的ATP;(4)根据收集的实验和比较基因组数据捕获生物体的已知代谢特征。补充表 2 总结了测试套件测试的所有功能。
为了保持一致性,现有的 818 AGORA1.03 重建(v.25.02.2019,可在 https://www.vmh.life/files/reconstructions/AGORA/1.03/AGORA-1.03.zip 上获得)也通过 DEMETER 进行了改进。去除了中间葡萄球菌 ATCC 27335 的 AGORA1.03 重建,因为它是新重建的中间链球菌 ATCC 27335 的副本。更改了8个AGORA1.03重建的名称,以正确的菌株测定和/或拼写(补充表1)。
DEMETER 已在 COBRA 工具箱中实现47并在 MATLAB (MathWorks) v.R2020b 中运行。
生成质量控制报告
使用 COBRA 工具箱中的 MetaboReport 工具确定每次 AGORA2 重建的质量控制报告和相关分数47.包含的质量检查与 Memote 一致42检查,分数的计算也是如此。所有 7,302 份报告都可以通过 https://metaboreport.live 访问。
药物反应的配方
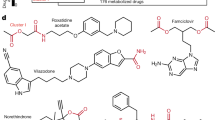
对已知可转化、降解、激活、失活或间接影响常用处方药的微生物酶进行了文献检索,总共产生了 15 种酶(图 1)。3a 和补充表 5),它们由 25 个基因编码(补充表 3b)。为了能够进行比较基因组分析,只考虑了可以与特定蛋白质编码基因相关的药物转化。如上所述,酶编码基因在其基因组环境中进行了分析,如参考文献所述。76使用 PubSEED 子系统26,53.从参考文献中检索到有关分析基因存在的其他信息。39,77,78.
对常用处方人类靶向药物的代谢命运进行了文献和数据库检索。从73篇同行评审论文中检索到287种药物代谢物和药物降解产物的结构,HMDB79, 药品银行79和 Transformer 数据库80.根据收集到的实验确定的药物结构、药物下游产物代谢物结构和反应机理配制反应。细胞溶质和细胞外酶促反应均根据鉴定的亚细胞蛋白位置进行配制。由于至少有六种在人体内进行葡萄糖醛酸化的药物已被证明是微生物β-葡萄糖醛酸酶的底物81,82(补充表6),假设所有回收到的葡萄糖醛酸化药物代谢物(共118种)都可以作为底物。此外,还配制了 β-葡萄糖醛酸酶反应,用于 33 种葡萄糖醛酸化药物代谢物,这些药物代谢物来自先前重建的人类药物代谢模块83和来自 Recon3D 的三种葡萄糖醛酸化激素(参考文献)。21).新的代谢物和反应按照用于 COBRA 重建的命名标准分配 VMH ID9,并在通过重建工具rBioNet确保质量和电荷平衡的同时进行配制75.总共有98种药物(图1)。3b)、353 种独特代谢物、381 种酶促反应、373 种交换反应和 710 种转运反应(附表 6a、b)。
原子-原子映射
COBRA 工具箱47函数“generateChemicalDatabase”用于生成原子-原子映射。获得AGORA2重建的原子-原子映射的过程可以总结如下:(1)AGORA2重建的3,533个代谢结构中的1,894个是从与其代谢物和不同化学数据库(如VMH)相关的SMILES和InChI中收集的23, 凯格74、HMDB79, PubChem84和 ChEBI85数据库;基于InChI算法对代谢结构进行标准化86并且可以在 VMH 数据库中找到23;(2)使用AGORA2重建中的标准化代谢物和反应化学计量法生成7,300个MDL RXN文件中的5,583个;(3) 7,300 个 AGORA2 反应中有 5,583 个使用反应解码器工具算法进行原子映射87用于主动转运反应和自定义算法47用于被动转运反应和偶联转运反应。可以在 VMH 数据库中找到原子-原子映射23并在 https://github.com/opencobra/ctf 免费提供。
模拟
所有仿真均在 MATLAB (MathWorks) v.R2020b 中执行,其中 IBM CPLEX (IBM) 作为线性和二次规划求解器。计算是在具有 2.80 GHz 处理器和 64 GB RAM 的塔上进行的,其中 12 个内核专用于并行化。使用COBRA工具箱中实现的功能进行模拟47.通量平衡分析(FBA)34用于模拟代谢通量。用于数据生成、数据分析和数据可视化的所有其他脚本均可在 https://github.com/ThieleLab/CodeBase 上获得。
重建资源的回收
与AGORA2相比,手动和半自动策划的重建检索如下:从BiGG数据库下载了72个完全手动策划的重建28 (http://bigg.ucsd.edu/)。通过gapseq生成的重建18(总共 8,075 个)从 ftp://ftp.rz.uni-kiel.de/pub/medsystbio/models/EnzymaticDataTestModels.zip 下载,并使用自定义脚本通过 R 中的 sybilSBML 包以 SBML 格式导出。岩浆17重建(共 1,333 个)是从 https://www.microbiomeatlas.org/data/MSP_GEM_models.zip 下载的。为了实现与AGORA2的可比性,所有检索到的重建中的交换反应都被翻译成VMH23通过自定义 MATLAB 脚本进行命名。此外,如果尚未存在,则添加ATP需求反应(VMH反应ID:DM_atp_c_),并以其他方式转换为VMH命名法。
通过 CarveMe 生成重建
从 NCBI (https://www.ncbi.nlm.nih.gov/assembly) 或 ENA (https://www.ebi.ac.uk/ena) 下载对应于 7,279 个 AGORA2 菌株的蛋白质 fasta 文件,随后用于运行 CarveMe。其余 23 株 AGORA2 菌株被排除在外,因为没有相应的蛋白质 FASTA 文件。使用 CarveMe 生成了 7,279 个菌株的重建15Python 3.7.13 上的 v.1.5.1(取自 https://www.python.org/downloads/release/python-3713)并依赖于 DIAMOND880.9.14 版。
通过gapseq生成重建
如上所述检索到的基因组 FASTA 文件被用作 gapseq 的输入18.使用 gapseq 1.2 共生成了 1,767 个模型,该模型在 R 中运行89v.4.1.2 在 Ubuntu 22.04 机器上。GLPK(Rglpk包)的R接口用作线性规划求解器。
通量和化学计量一致的反应
通量和化学计量一致反应的子集,如参考文献中所定义。29,通过 COBRA 工具箱中实现的“findFluxConsistentSubset”和“findStoichConsistentSubset”函数检索47.随后,对于每个 AGORA2 重建和相应的 KBase 草图重建以及通过 CarveMe 生成的 5,587 个重建,确定了化学计量和通量一致反应的比例,不包括交换和需求反应15,通过 gapseq 生成了 8,075 个重建18, 1,333 岩浆17来自 BiGG 数据库的重建和 73 个精选重建28.简而言之,重建中化学计量一致的反应子集包括所有质量和电荷守恒的反应,不包括交换、需求和汇反应,根据定义,这些反应是质量和电荷不平衡的29.通量一致反应的子集由所有反应组成,这些反应可以在定义的约束集下携带通量29.
针对三个独立的实验数据集进行验证
为了独立评估基因组规模重建的预测潜力,从三个来源检索了关于代谢物摄取和分泌的独立(即不用于重建过程)实验数据30,32,33并映射到 VMH23通过自定义 MATLAB 脚本进行命名。实验数据包括来自NJC19资源的AGORA2中457个物种(5,341个菌株)和269个代谢物的物种水平正负代谢物摄取和分泌数据30,以及来自参考文献的物种水平正代谢物摄取数据。32AGORA2 中有 184 种(328 株)和 85 种代谢物。此外,从BacDive数据库中检索了676株AGORA2菌株和220株代谢物的菌株分辨阳性和阴性代谢物摄取和分泌数据,以及881株AGORA2菌株和31种酶的阳性和阴性酶活性数据33.酶数据被映射到每个比较重建资源命名空间中的相应反应。阳性数据表明微生物的代谢物摄取、分泌能力或酶活性已得到证实,而阴性数据表明该微生物不具备这种能力。对于每个检索到的正数据点或负数据点,使用FBA在无限培养基上分别通过最小化或最大化相应的交换反应来计算相应模型吸收或产生相应代谢物的能力。对于酶数据,测试了模型中是否存在至少一个映射到相应酶的反应,并且可以携带非零通量。如果数据点为正,并且相应的模型也可以通过相应的酶促反应吸收或分泌代谢物或产生通量,则会导致真阳性预测,而当已知微生物具有这种能力时,就会发生假阴性预测,但相应的模型没有捕获该性状。如果数据点为负,并且相应的模型也无法吸收或分泌代谢物,或者没有通过映射到酶的任何反应产生通量,则会导致真正的阴性预测,否则预测为假阳性。
计算了三个实验数据集的预测精度。为了评估 AGORA2 与其他重建资源相比的预测潜力,对 KBase 草案重建中的菌株重复分析;CarveMe重建;以及BiGG、gapseq和MAGMA重建,这些重建与AGORA2生物体重叠,有可用数据。为此,通过混合效应逻辑回归测试了所有资源的预测价值,计算机预测作为预测变量,体内行为(二元)作为响应变量,同时引入模型作为随机效应变量,考虑了来自同一模型的不同代谢物预测的随机依赖性。此外,计算了所有资源的每个模型的准确度,然后通过非参数符号秩测试与 AGORA2 精度进行比较。根据三个数据集测试的比较重建资源中所有菌株的列表显示在补充表4a中。所有脚本均可在 https://github.com/ThieleLab/CodeBase 获得。
根据独立实验数据验证药物代谢能力
对体外实验进行了文献检索,证明了人类微生物菌株通过 15 种带注释的酶代谢重建药物的能力,产生了 253 个药物-微生物对(补充表 7)。由于该数据包含正数据和负数据,因此可能会出现如上所述的真阳性、真阴性、假阳性和假阴性预测。如果没有发现关于该酶的特定重建药物的研究,则检索有关该酶一般活性的研究。如果可能,将测试的微生物与菌株水平上的AGORA2模型相匹配,否则使用泛物种模型。随后,通过计算相应的反应是否可以携带通量来测试具有可用数据的 164 个 AGORA2 模型(补充表 7)通过各自酶代谢药物的能力。在确定真阳性、真阴性、假阳性和假阴性预测的数量后计算预测的准确性、敏感性和特异性。P值通过Fisher精确检验计算,对于敏感性分析,通过混合效应逻辑回归计算,包括模型作为随机效应变量,考虑来自同一模型的预测的随机依赖性。
药物产量
为了确定每种菌株的药物代谢能力,所有AGORA2菌株都受到模拟西方饮食的约束20使用FBA最小化通过对应于每种药物的交换反应的通量,对应于药物的最大摄取率。对于所有能够吸收至少一种药物的AGORA2生物体,每克1毫摩尔药物的ATP,碳和氨的产量干重每小时评估如下。每次重建都被限制为只允许吸收水、磷酸盐和氧气(VMH ID:h2o、pi、o2)。氨和一氧化碳的需求反应2添加了丙酮酸(作为碳源的替代物)(VMH ID:nh4,co2,pyr),而在每次重建中都已经存在对ATP的需求反应(VMH ID:atp)。接下来,以每克1mmol的摄取率逐个摄取每种药物代谢物(总共15个,每种酶一个代表)干重每小时。对于每种药物代谢物,ATP、氨、CO 的产率2和来自每种药物代谢物的丙酮酸通过FBA通过通过各自的需求反应最大化通量来计算。作为对照,还计算了每克 1 毫米的产量干重每小时葡萄糖,不添加任何代谢物。
通过单个肠道微生物组模拟药物代谢
此前,已经对616名日本结直肠癌患者和健康对照者的粪便样本进行了宏基因组测序38.该队列的物种水平丰度,已用 MetaPhIAn2 确定(参考文献)。90),从 https://www.nature.com/articles/s41591-019-0458-7#MOESM3 中检索。物种水平上的未分类分类群、真核生物和病毒被排除在外。在剩下的517个物种中,501个(97%)可以映射到1,738个AGORA2物种上。AGORA2 的泛物种模型是通过“createPanModels”函数创建的。从泛物种模型中,通过计算高效的管道构建了 616 个样本中每个样本的个性化微生物组模型43以物种水平的丰度作为输入数据,并按其他说明进行参数化10,60.对于每个人,我们将样品中丰度为非零的所有微生物模型整合到一个个性化的微生物组模型中。为了将模型与适当的饮食约束联系起来,前面描述的模拟日本平均饮食41(补充表12)被使用。为了预测每种微生物组的药物转化潜力,使用FBA对13种药物代谢终产物的粪便分泌反应进行逐一优化34,同时以每克 1,000 毫摩尔的事实上无限的摄取率提供相应的前体药物和氧气干重每小时。
影子价格分析
为了确定微生物组模型中对微生物组代谢药物的综合潜力很重要的物种,如前所述进行了影子价格分析60.简而言之,影子价格是每个 FBA 解决方案的一个特征(即影子价格是原始线性规划问题的对偶),它反映了模型中每个代谢物通过目标函数对通量的贡献8.代谢物的非零影子价格表明,该代谢物通过优化的目标函数(即在我们的例子中,药物代谢产物的分泌)对总通量容量具有重要意义。影子价格为零表示增加该代谢物的可用性不会改变通过目标函数的通量。为了确定每个微生物组模型中 13 种药物转换潜力的瓶颈物种,检索了物种生物质代谢物 ('species_biomass[c]') 的非零影子价格,该价格反映了物种对群落生物质反应的贡献。
统计分析
我们统计分析了13种药物代谢物的净生产能力(图1)。6a) 在 252 例健康个体和 364 例 CRC 患者中。对于每种药物代谢物,我们计算了通量大于零的平均通量和微生物组的份额。药物代谢物在超过50%的病例中通量为零,被二分法(可以产生与不能产生),随后通过逻辑回归进行分析。使用异方差稳健标准误差通过线性回归分析非零条目超过 50% 的药物代谢物。首先,我们通过广义线性回归(逻辑或线性)研究了基本协变量(年龄、性别和 BMI)的潜在影响,净生产能力是响应变量(二分法或度量)。年龄和 BMI 作为限制三次样条引入模型91使用四个节(5% 百分位数、33% 百分位数、66% 百分位数和 95% 百分位数)产生三个样条变量,每个样条变量都用于测试潜在的非线性关系。然后通过 Wald 检验同时在零上测试属于年龄(或分别为 BMI)的三个样条变量来确定显着性91.虽然在年龄方面发现了大量的非线性,但无法确定非线性 BMI 效应的迹象。因此,最终模型仅包括线性 BMI 项。其次,我们测试了净生产能力与病例控制状态的潜在关联。该测试是通过广义线性回归(逻辑或线性)完成的,净生产能力是响应变量(二分法或公制),同时调整年龄(限制三次样条)、性别(男性/女性)和 BMI(线性)。我们使用 FDR 校正了多个测试,调整了每个分析流 13 个测试的显著性值。当 P < 为 0.05 时,该检验被认为是标称显著的,如果 FDR < 为 0.05,则 FDR 校正显著。对于敏感性分析,我们使用平均欧洲饮食而不是日本饮食重新计算了药物代谢能力。然后,我们计算了日本饮食和欧洲平均饮食下分泌电位之间每种药物代谢物的 Pearson 相关性。所有统计分析均使用STATA 17/MP进行。所有脚本均可在 https://github.com/ThieleLab/CodeBase 获得。
使用基于 AGORA2 的群落模型预测粪便代谢物-物种关联的标志
我们利用了文献中公开可用的代谢组数据集(n = 347)。38.为了测试基于 AGORA2 的群落建模是否能够预测 CRC 样品中物种存在与粪便代谢物浓度之间的统计关联迹象,我们用粪便代谢组数据计算了 52 种代谢物的最大净分泌量,其中超过 50% 的样品浓度高于检测限。使用微生物组建模工具箱中的 mgPipe 模块计算代谢物净分泌10,43同时依靠计算高效的通量变异性分析92.然后,我们计算了至少 10% 的微生物组和最多 90% 的微生物组中存在的每个物种的物种(二元预测因子:存在的物种与不存在的物种)对多变量回归中每个粪便代谢物浓度的影响,调整年龄、性别、BMI 和研究组。然后,我们过滤了所有物种代谢物的关联,P<为0.05。接下来,我们计算了物种存在对相应代谢物群落净分泌的影响。最后,我们计算了每种代谢物体内关联统计量和计算机结合统计量之间的符号一致性。显著性由 Fisher 精确检验确定,并应用 FDR 校正,占 52 个检验。请注意,应谨慎对待 P 值,因为由于代谢组和微生物组数据的多变量性质,各种关联统计量的迹象可能会聚集在一起。
数据可视化
AGORA2生物的系统发育树在PhyloT(https://phylot.biobyte.de/)中构建,并在iTOL中可视化(https://itol.embl.de/)93.小提琴图是在 BoxPlotR (http://shiny.chemgrid.org/boxplotr/) 中生成的。通过t分布随机邻域嵌入(t-SNE)按反应存在聚类52在 MATLAB 中使用 t-SNE 实现执行,欧几里得距离,将 barneshut 设置为算法,并将困惑度设置为 30。在所有聚类菌株中代表性小于0.5%的分类单元被排除在t-SNE图中。通过Kruskal-Wallis检验确定不同分类单元之间坐标差异的显著性。圆图是使用 Circos 的在线实现生成的94.图6和补充图。9 个是使用 STATA 16/MP 的图形功能生成的。所有其他数据均在MATLAB和R中可视化89.
报告摘要
有关研究设计的更多信息,请参阅本文链接的《自然投资组合报告摘要》。
数据可用性
7,302 个 AGORA2 重建可在 https://www.vmh.life/ 免费获得(https://www.vmh.life/files/reconstructions/AGORA2 可批量下载)。所有重建的质量控制报告均可在 https://metaboreport.live/ 获得。
代码可用性
https://github.com/ThieleLab/CodeBase 提供编码和输入数据,以重现AGORA2重建和微生物组模型的生成,以及所有模拟和分析。
引用
林奇,SV和佩德森,O.健康和疾病中的人体肠道微生物组。N. Engl. J. Med. 375, 2369–2379 (2016).
Nebert, D. W., Zhang, G. & Vesell, E. S.从人类遗传学和基因组学到药物遗传学和药物基因组学:过去的经验教训,未来的方向。药物代谢修订版 40, 187–224 (2008)。
Tralau, T., Sowada, J. & Luch, A. 关于人类微生物组及其异生代谢的见解:对其对人类生理学的影响了解多少?专家意见。药物Metab。毒理学。 11, 411–425 (2015).
Spanogiannopoulos,P.,Bess,E.N.,Carmody,RN和Turnbaugh,PJ我们体内的微生物药剂师:异源代谢的宏基因组学观点。Nat. Rev. 微生物学。 14, 273–287 (2016).
Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R. & Goodman, A. L. 通过肠道细菌及其基因绘制人类微生物组药物代谢图谱。自然 570, 462–467 (2019)。
Javdan, B. et al. Personalized mapping of drug metabolism by the human gut microbiome. Cell 181, 1661–1679 e1622 (2020).
Guthrie, L. & Kelly, L. Bringing microbiome-drug interaction research into the clinic. EBioMedicine 44, 708–715 (2019).
Palsson, B. Systems Biology: Properties of Reconstructed Networks (Cambridge Univ. Press, 2006).
Thiele, I. & Palsson, B. Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121 (2010).
Baldini, F. et al. The Microbiome Modeling Toolbox: from microbial interactions to personalized microbial communities. Bioinformatics 35, 2332–2334 (2019).
Diener, C., Gibbons, S. M. & Resendis-Antonio, O. MICOM: metagenome-scale modeling to infer metabolic interactions in the gut microbiota. mSystems 5, e00606–e00619 (2020).
Magnusdottir, S. & Thiele, I. Modeling metabolism of the human gut microbiome. Curr. Opin. Biotechnol. 51, 90–96 (2018).
van der Ark, K. C. H., van Heck, R. G. A., Martins Dos Santos, V. A. P., Belzer, C. & de Vos, W. M. More than just a gut feeling: constraint-based genome-scale metabolic models for predicting functions of human intestinal microbes. Microbiome 5, 78 (2017).
Lagier, J. C. et al. Many more microbes in humans: enlarging the microbiome repertoire. Clin. Infect. Dis. 65, S20–S29 (2017).
Machado, D., Andrejev, S., Tramontano, M. & Patil, K. R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 46, 7542–7553 (2018).
Zorrilla, F., Buric, F., Patil, K. R. & Zelezniak, A. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes. Nucleic Acids Res. 49 (2021). https://doi.org/10.1093/nar/gkab 815
Bidkhori, G. et al. The reactobiome unravels a new paradigm in human gut microbiome metabolism. Preprint at https://doi.org/10.1101/2021.02.01.428114 (2021).
Zimmermann, J., Kaleta, C. & Waschina, S. gapseq: informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biol. 22, 81 (2021).
Heinken, A., Magnusdottir, S., Fleming, R. M. T. & Thiele, I. DEMETER: efficient simultaneous curation of genome-scale reconstructions guided by experimental data and refined gene annotations. Bioinformatics 37, 3974–3975 (2021).
Magnusdottir, S. et al. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nat. Biotechnol. 35, 81–89 (2017).
Brunk, E. et al. Recon3D enables a three-dimensional view of gene variation in human metabolism. Nat. Biotechnol. 36, 272–281 (2018).
Thiele, I. et al. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Mol. Syst. Biol. 16, e8982 (2020).
Noronha, A. et al. The Virtual Metabolic Human database: integrating human and gut microbiome metabolism with nutrition and disease. Nucleic Acids Res. 47, D614–D624 (2019).
Arkin, A. P. et al. KBase: the United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 36, 566–569 (2018).
Bernstein, D. B., Sulheim, S., Almaas, E. & Segre, D. Addressing uncertainty in genome-scale metabolic model reconstruction and analysis. Genome Biol. 22, 64 (2021).
Aziz, R. K. et al. SEED servers: high-performance access to the SEED genomes, annotations, and metabolic models. PLoS ONE 7, e48053 (2012).
Henry, C. S. et al. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982 (2010).
Norsigian, C. J. et al. BiGG Models 2020: multi-strain genome-scale models and expansion across the phylogenetic tree. Nucleic Acids Res. 48, D402–D406 (2020).
Fleming, R. M., Vlassis, N., Thiele, I. & Saunders, M. A. Conditions for duality between fluxes and concentrations in biochemical networks. J. Theor. Biol. 409, 1–10 (2016).
Lim, R. et al. Large-scale metabolic interaction network of the mouse and human gut microbiota. Sci. Data 7, 204 (2020).
Sung, J. et al. Global metabolic interaction network of the human gut microbiota for context-specific community-scale analysis. Nat. Commun. 8, 15393 (2017).
Madin, J. S. et al. A synthesis of bacterial and archaeal phenotypic trait data. Sci. Data 7, 170 (2020).
Reimer, L. C. et al. BacDive in 2019: bacterial phenotypic data for high-throughput biodiversity analysis. Nucleic Acids Res. 47, D631–D636 (2019).
Orth, J. D., Thiele, I. & Palsson, B. O. What is flux balance analysis? Nat. Biotechnol. 28, 245–248 (2010).
Zimmermann, M., Zimmermann-Kogadeeva, M., Wegmann, R. & Goodman, A. L. Separating host and microbiome contributions to drug pharmacokinetics and toxicity. Science 363, eaat9931 (2019).
Pollet, R. M. et al. An atlas of β-glucuronidases in the human intestinal microbiome. Structure 25, 967–977.e5 (2017).
Heinken, A., Hertel, J. & Thiele, I. Metabolic modelling reveals broad changes in gut microbial metabolism in inflammatory bowel disease patients with dysbiosis. Syst. Biol. Appl. 7, 19 (2021).
Yachida, S. et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 25, 968–976 (2019).
Maini Rekdal, V., Bess, E. N., Bisanz, J. E., Turnbaugh, P. J. & Balskus, E. P. Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323 (2019).
Wirbel, J. et al. Meta-analysis of fecal metagenomes reveals global microbial signatures that are specific for colorectal cancer. Nat. Med. 25, 679–689 (2019).
Hertel, J., Heinken, A., Martinelli, F. & Thiele, I. Integration of constraint-based modeling with fecal metabolomics reveals large deleterious effects of Fusobacterium spp. on community butyrate production. Gut Microbes 13, 1–23 (2021).
Lieven, C. et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 38, 272–276 (2020).
Heinken, A. & Thiele, I. Microbiome Modelling Toolbox 2.0: efficient, tractable modelling of microbiome communities. Bioinformatics 38, 2367–2368 (2022).
Sen, P. & Oresic, M. Metabolic modeling of human gut microbiota on a genome scale: an overview. Metabolites 9, 22 (2019).
Monk, J. M. et al. iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 35, 904–908 (2017).
Heinken, A., Basile, A. & Thiele, I. Advances in constraint-based modelling of microbial communities. Curr. Opin. Syst. Biol. 27 (2021). https://doi.org/10.1016/j.coisb.2021.05.007
Heirendt, L. et al. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nat. Protoc. 14, 639–702 (2019).
Bebb, J. R. & Scott, B. B. How effective are the usual treatments for ulcerative colitis? Aliment. Pharm. Ther. 20, 143–149 (2004).
Thiele, I., Clancy, C. M., Heinken, A. & Fleming, R. M. T. Quantitative systems pharmacology and the personalized drug-microbiota-diet axis. Curr. Opin. Syst. Biol. 4, 43–52 (2017).
Krauss, M. et al. Integrating cellular metabolism into a multiscale whole-body model. PLoS Comput. Biol. 8, e1002750 (2012).
Heinken, A., Basile, A., Hertel, J., Thinnes, C. & Thiele, I. Genome-scale metabolic modeling of the human microbiome in the era of personalized medicine. Annu. Rev. Microbiol. 75, 199–222 (2021).
van der Maaten, L. & Hinton, G. Viualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Overbeek, R. et al. The subsystems approach to genome annotation and its use in the Project to Annotate 1000 Genomes. Nucleic Acids Res. 33, 5691–5702 (2005).
Bisanz, J. E. et al. A genomic toolkit for the mechanistic dissection of intractable human gut bacteria. Cell Host Microbe 27, 1001–1013.e9 (2020).
Forster, S. C. et al. A human gut bacterial genome and culture collection for improved metagenomic analyses. Nat. Biotechnol. 37, 186–192 (2019).
Disz, T. et al. Accessing the SEED genome databases via Web services API: tools for programmers. BMC Bioinformatics 11, 319 (2010).
Ravcheev, D. A. & Thiele, I. Systematic genomic analysis reveals the complementary aerobic and anaerobic respiration capacities of the human gut microbiota. Front. Microbiol. 5, 674 (2014).
Magnusdottir, S., Ravcheev, D., de Crecy-Lagard, V. & Thiele, I. Systematic genome assessment of B-vitamin biosynthesis suggests co-operation among gut microbes. Front. Genet. 6, 148 (2015).
Ravcheev, D. A. & Thiele, I. Genomic analysis of the human gut microbiome suggests novel enzymes involved in quinone biosynthesis. Front. Microbiol. 7, 128 (2016).
Heinken, A. et al. Personalized modeling of the human gut microbiome reveals distinct bile acid deconjugation and biotransformation potential in healthy and IBD individuals. Microbiome 7, 75 (2019).
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y. & Morishima, K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361 (2017).
Wolf, Y. I. & Koonin, E. V. A tight link between orthologs and bidirectional best hits in bacterial and archaeal genomes. Genome Biol. Evol. 4, 1286–1294 (2012).
Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Marchler-Bauer, A. et al. CDD: conserved domains and protein three-dimensional structure. Nucleic Acids Res. 41, D348–D352 (2013).
Yu, C. S., Chen, Y. C., Lu, C. H. & Hwang, J. K. Prediction of protein subcellular localization. Proteins 64, 643–651 (2006).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797 (2004).
Larkin, M. A. et al. Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007).
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321 (2010).
Huson, D. H. et al. Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 8, 460 (2007).
Krieg, N. et al. Bergey’s Manual® of Systematic Bacteriology (Springer, New York, 2010).
Chen, I. A. et al. IMG/M v.5.0: an integrated data management and comparative analysis system for microbial genomes and microbiomes. Nucleic Acids Res. 47, D666–D677 (2019).
Aziz, R. K. et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9, 75 (2008).
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K. & Tanabe, M. New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47, D590–D595 (2019).
Thorleifsson, S. G. & Thiele, I. rBioNet: a COBRA toolbox extension for reconstructing high-quality biochemical networks. Bioinformatics 27, 2009–2010 (2011).
Osterman, A. & Overbeek, R. Missing genes in metabolic pathways: a comparative genomics approach. Curr. Opin. Chem. Biol. 7, 238–251 (2003).
Zou, L. et al. Bacterial metabolism rescues the inhibition of intestinal drug absorption by food and drug additives. Proc. Natl Acad. Sci. USA 117, 16009–16018 (2020).
Koppel, N., Bisanz, J. E., Pandelia, M. E., Turnbaugh, P. J. & Balskus, E. P. Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. eLife 7, e33953 (2018).
Wishart, D. S. et al. HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 46, D608–D617 (2018).
Hoffmann, M. F. et al. The Transformer database: biotransformation of xenobiotics. Nucleic Acids Res. 42, D1113–D1117 (2014).
Wallace, B. D. et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science 330, 831–835 (2010).
Saitta, K. S. et al. Bacterial β-glucuronidase inhibition protects mice against enteropathy induced by indomethacin, ketoprofen or diclofenac: mode of action and pharmacokinetics. Xenobiotica 44, 28–35 (2014).
Sahoo, S., Haraldsdottir, H., Fleming, R. M. & Thiele, I. Modeling the effects of commonly used drugs on human metabolism. FEBS J. 282, 297–317 (2015).
Kim, S. et al. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. 47, D1102–D1109 (2019).
Hastings, J. et al. The ChEBI reference database and ontology for biologically relevant chemistry: enhancements for 2013. Nucleic Acids Res. 41, D456–D463 (2013).
Heller, S. R., McNaught, A., Pletnev, I., Stein, S. & Tchekhovskoi, D. InChI, the IUPAC international chemical identifier. J. Cheminform 7, 23 (2015).
Rahman, S. A. et al. Reaction Decoder Tool (RDT): extracting features from chemical reactions. Bioinformatics 32, 2065–2066 (2016).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60 (2015).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2013).
Truong, D. T. et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12, 902–903 (2015).
Harrell, F. E. Regression Modeling Strategies: with Applications to Linear Models, Logistic Regression, and Survival Analysis (Springer, 2001).
Gudmundsson, S. & Thiele, I. Computationally efficient flux variability analysis. BMC Bioinformatics 11, 489 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259 (2019).
Krzywinski, M. et al. Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645 (2009).
Acknowledgements
We thank P. Turnbaugh for providing genome sequences for 26 Eggerthella lenta strains, J. Krumsiek for communicating mouse-associated microbial species, C. Thinnes for valuable discussions, and L. Moussu and S. Smajic for their help with the comparative genomic effort. This study was funded by grants from the European Research Council under the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 757922) to I.T., from the National Institute on Aging (grants no. 1RF1AG058942 and no. 1U19AG063744), from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie Actions (grant agreement no. 859890), from the Marine Institute (Grant-Aid Agreement No. PBA/BIO/20/03) and Science Foundation Ireland funded by the Irish Government under the BlueBio ERA-NET Co-fund (H2020 Project number 817992) and from the Science Foundation Ireland under grant no. 12/RC/2273-P2.
Author information
Authors and Affiliations
Contributions
I.T. and A.H. conceived the study. D.A.R., G.A. and O.E.O. performed comparative genomic analyses. I.T., A.H., S.M., M.H., F.M., J.N.E. and C.S.H. created KBase draft reconstructions. A.H. and S.M. built the semiautomated reconstruction pipeline and the test suite. G.A. and A.H. translated reaction and metabolite identifiers to VMH nomenclature. M.H., G.A., A.H. and F.M. collected experimental data. M.H., F.M. and A.H. collected organism information. M.N. and A.H. formulated the drug reactions. A.H. performed continuous reconstruction testing and curation. A.H. performed simulations. A.H. and J.H. analyzed and visualized the data. J.H. performed statistical analyses. B.N. built CarveMe and gapseq reconstructions. G.P. and R.M.T.F. performed atom–atom mappings. A.H. and J.H. drafted the paper. All authors edited the paper. I.T. supervised the study.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Biotechnology thanks Matej Orešič and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary Information
Supplementary Notes 1–11 and Figs. 1–10.
Supplementary Data 1
Supplementary Tables 1–12.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Heinken, A., Hertel, J., Acharya, G. et al. Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nat Biotechnol 41, 1320–1331 (2023). https://doi.org/10.1038/s41587-022-01628-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41587-022-01628-0
Subjects
This article is cited by
-
Predicting microbial interactions with approaches based on flux balance analysis: an evaluation
BMC Bioinformatics (2024)
-
Flux sampling in genome-scale metabolic modeling of microbial communities
BMC Bioinformatics (2024)
-
Long-term relapse-free survival enabled by integrating targeted antibacteria in antitumor treatment
Nature Communications (2024)
-
Utilization of the microbiome in personalized medicine
Nature Reviews Microbiology (2024)
-
High-throughput transcriptomics of 409 bacteria–drug pairs reveals drivers of gut microbiota perturbation
Nature Microbiology (2024)