
Abstract 抽象的
Glioblastoma (GBM) is the most common malignant tumor in the brain with temozolomide (TMZ) as the only approved chemotherapy agent. GBM is characterized by susceptibility to radiation and chemotherapy resistance and recurrence as well as low immunological response. There is an urgent need for new therapy to improve the outcome of GBM patients. We previously reported that 3-O-acetyl-11-keto-β-boswellic acid (AKBA) inhibited the growth of GBM. In this study we characterized the anti-GBM effect of S670, a synthesized amide derivative of AKBA, and investigated the underlying mechanisms. We showed that S670 dose-dependently inhibited the proliferation of human GBM cell lines U87 and U251 with IC50 values of around 6 μM. Furthermore, we found that S670 (6 μM) markedly stimulated mitochondrial ROS generation and induced ferroptosis in the GBM cells. Moreover, S670 treatment induced ROS-mediated Nrf2 activation and TFEB nuclear translocation, promoting protective autophagosome and lysosome biogenesis in the GBM cells. On the other hand, S670 treatment significantly inhibited the expression of SXT17, thus impairing autophagosome-lysosome fusion and blocking autophagy flux, which exacerbated ROS accumulation and enhanced ferroptosis in the GBM cells. Administration of S670 (50 mg·kg−1·d−1, i.g.) for 12 days in a U87 mouse xenograft model significantly inhibited tumor growth with reduced Ki67 expression and increased LC3 and LAMP2 expression in the tumor tissues. Taken together, S670 induces ferroptosis by generating ROS and inhibiting STX17-mediated fusion of autophagosome and lysosome in GBM cells. S670 could serve as a drug candidate for the treatment of GBM.
胶质母细胞瘤(GBM)是脑部最常见的恶性肿瘤,替莫唑胺(TMZ)是唯一批准的化疗药物。 GBM 的特点是易受放疗和化疗耐药和复发以及免疫反应低。迫切需要新的疗法来改善 GBM 患者的预后。我们之前报道过 3-O-乙酰基-11-酮-β-乳香酸 (AKBA) 抑制 GBM 的生长。在本研究中,我们表征了 S670(AKBA 的合成酰胺衍生物)的抗 GBM 作用,并研究了其潜在机制。我们发现,S670 剂量依赖性地抑制人 GBM 细胞系 U87 和 U251 的增殖,IC 50值约为 6 μM。此外,我们发现 S670 (6 μM) 显着刺激 GBM 细胞中线粒体 ROS 的产生并诱导铁死亡。此外,S670 处理诱导 ROS 介导的 Nrf2 激活和 TFEB 核转位,促进 GBM 细胞中的保护性自噬体和溶酶体生物发生。另一方面,S670处理显着抑制SXT17的表达,从而损害自噬体-溶酶体融合并阻断自噬流,从而加剧GBM细胞中ROS的积累并增强铁死亡。在U87小鼠异种移植模型中施用S670(50 mg·kg -1 ·d -1 ,ig)12天显着抑制肿瘤生长,肿瘤组织中Ki67表达减少,LC3和LAMP2表达增加。综上所述,S670 通过产生 ROS 并抑制 GBM 细胞中 STX17 介导的自噬体和溶酶体融合来诱导铁死亡。 S670可以作为治疗GBM的候选药物。
Similar content being viewed by others
其他人正在查看类似内容

Introduction 介绍
GBM is the most commonly occurring malignant primary brain tumor, which accounts for 48.6% of malignant cases in central nervous system (CNS) tumors [1]. GBM was classified as grade IV gliomas by World Health Organization (WHO), indicating the most malignant behavior [2]. Surgery followed by radiotherapy and further adjuvant temozolomide (TMZ) is the standard treatment for GBM patients. Although therapeutic advances have reached increasing improvements in shorter-term survival rates, GBM patients have remained a poor prognosis [3]. Due to the heterogeneity in the tumor microenvironment, infiltration of glioma stem cells and low immunogenicity, GBM is characterized by susceptibility to radiation and chemotherapy resistance and recurrence, and low immunological response [4]. Thus, there is an urgent need for new effective therapy to improve the outcome of GBM patients.
GBM是最常见的恶性原发性脑肿瘤,占中枢神经系统(CNS)肿瘤恶性病例的48.6%[ 1 ]。 GBM被世界卫生组织(WHO)列为IV级胶质瘤,恶性程度最高[ 2 ]。手术后放疗和进一步辅助替莫唑胺 (TMZ) 是 GBM 患者的标准治疗方法。尽管治疗进展已使短期生存率不断提高,但 GBM 患者的预后仍然较差[ 3 ]。由于肿瘤微环境的异质性、胶质瘤干细胞的浸润和低免疫原性,GBM具有易发生放化疗耐药和复发、免疫应答低等特点[ 4 ]。因此,迫切需要新的有效疗法来改善 GBM 患者的预后。
Ferroptosis, a type of cell death characterized by iron-dependent lipid peroxidation, is implicated in a wide variety of diseases including cancers [5]. Ferroptosis was found as the major programmed cell death (PCD) process in GBM and was associated with immunosuppression [6]. Emerging molecular regulators of ferroptosis were identified in GBM and have been comprehensively reviewed by Shi et al. [7]. Targeting ferroptosis provides new opportunities for the treatment of GBM. Apatinib was found to induce ferroptosis in glioma via modulating VEGFR2/Nrf2 Pathway [8]. Natural products are the main sources of new drugs [9]. A series of molecules from natural products exerted anti-GBM activity by inducing ferroptosis such as brucine [10], plumbagin [11], and pseudolaric acid B [12]. Moreover, targeting ferroptosis can even overcome temozolomide resistance in GBM [13].
铁死亡是一种以铁依赖性脂质过氧化为特征的细胞死亡,与包括癌症在内的多种疾病有关[ 5 ]。铁死亡被发现是 GBM 中主要的程序性细胞死亡 (PCD) 过程,并且与免疫抑制相关 [ 6 ]。 Shi 等人在 GBM 中鉴定了铁死亡的新兴分子调节因子,并对其进行了全面综述。 [ 7 ]。靶向铁死亡为 GBM 的治疗提供了新的机会。阿帕替尼被发现通过调节 VEGFR2/Nrf2 通路诱导胶质瘤铁死亡[ 8 ]。天然产物是新药的主要来源[ 9 ]。一系列来自天然产物的分子通过诱导铁死亡而发挥抗GBM活性,例如马钱子碱[ 10 ]、白花丹素[ 11 ]和假月桂酸B[ 12 ]。此外,针对铁死亡甚至可以克服GBM中的替莫唑胺耐药性[ 13 ]。
Autophagy is an evolutionally conserved cellular process. Autophagosome sequesters damaged cellular contents and then fuses with the lysosome to produce autolysosome and autophagy substrates are degraded by hydrolases and recycled to sustain cellular metabolism [14]. Autophagy plays multiple functions in tumor growth. In oncogenesis, tumor-suppressive autophagy could maintain cellular and genomic homeostasis. However, in tumor progression, cytoprotective autophagy in tumor cells is boosted to encounter different cellular stress [15, 16]. In recent years, targeting autophagy has been proposed as a potential therapy for GBM. Inhibition of protective autophagy can sensitize GBM cells to chemotherapy agents or radiotherapy but excessive autophagy activation in GBM can also induce autophagic cell death [17]. A diversity of autophagy modulators including natural products, HDAC inhibitors, nanoparticles, etc [18] have been investigated to assess the anti-GBM effect.
自噬是进化上保守的细胞过程。自噬体隔离受损的细胞内容物,然后与溶酶体融合产生自噬溶酶体,自噬底物被水解酶降解并回收以维持细胞代谢[ 14 ]。自噬在肿瘤生长中发挥多种作用。在肿瘤发生过程中,肿瘤抑制自噬可以维持细胞和基因组稳态。然而,在肿瘤进展过程中,肿瘤细胞的细胞保护性自噬会增强,以应对不同的细胞应激[ 15 , 16 ]。近年来,靶向自噬被提出作为 GBM 的潜在治疗方法。抑制保护性自噬可以使 GBM 细胞对化疗药物或放疗敏感,但 GBM 中过度的自噬激活也可以诱导自噬细胞死亡 [ 17 ]。人们已经研究了多种自噬调节剂,包括天然产物、HDAC抑制剂、纳米颗粒等[ 18 ]来评估其抗GBM效果。
As one of the most active pentacyclic triterpene in resin/gum of Boswellia serrata, 3-O-acetyl-11-keto-β-boswellic acid (AKBA) exhibited potent anti-inflammatory activity via inhibiting the release of leukotriene, and the activity of cyclooxygenase 1/2 (COX1/2) and 5-lipoxygenase (5-LOX) [19, 20]. Recent studies also demonstrated that AKBA was used for the treatment of several CNS diseases including neuroprotection of ischemic brain injury, amelioration of neurodegenerative disease, remission of brain edema, and anti-glioma activity [21, 22]. Our group found that AKBA exhibited potent anti-GBM effects. AKBA could block cell cycle and induce apoptosis in GBM cells [23]. Moreover, AKBA can also inhibit tumor growth, ameliorate the aberrant metabolic landscape and inhibit autophagy in GBM [24]. Thus, AKBA could be a potential therapeutic agent for GBM. However, the limitation of IC50 confined the further application of AKBA in cancer treatment [23]. Therefore, we decided to modify structure of AKBA to improve the anti-GBM effect.
3-O-乙酰基-11-酮-β-乳香酸(AKBA)是乳香树树脂/树胶中最活跃的五环三萜之一,通过抑制白三烯的释放而表现出有效的抗炎活性,并且环氧合酶 1/2 (COX1/2) 和 5-脂氧合酶 (5-LOX) [ 19 , 20 ]。最近的研究还表明,AKBA 可用于治疗多种 CNS 疾病,包括缺血性脑损伤的神经保护、神经退行性疾病的改善、脑水肿的缓解和抗神经胶质瘤活性 [ 21 , 22 ]。我们的小组发现 AKBA 表现出有效的抗 GBM 作用。 AKBA可以阻断GBM细胞的细胞周期并诱导细胞凋亡[ 23 ]。此外,AKBA 还可以抑制 GBM 中的肿瘤生长、改善异常代谢景观并抑制自噬 [ 24 ]。因此,AKBA 可能是 GBM 的潜在治疗剂。然而,IC 50的限制限制了AKBA在癌症治疗中的进一步应用[ 23 ]。因此,我们决定修改AKBA的结构以提高抗GBM效果。
Here, a total of 23 novel amide derivatives of AKBA were designed and synthesized to enhance pharmacological efficacy, and compound S670 was found to exhibit much lower IC50 than AKBA [23]. S670 inhibited the proliferation of GBM cells in vitro. Following mechanism study revealed that S670 exerted anti-GBM activity via stimulating ROS generation to induce ferroptosis in GBM cells. Moreover, S670 could induce autophagosome biogenesis via ROS-activated Nrf2 nuclear translocation and stimulate lysosome biogenesis by ROS-activated TFEB nuclear translocation. However, S670 interrupted the fusion of autophagosome and lysosome to exacerbate ferroptosis by inhibiting the expression of STX17 in GBM cells. In addition, S670 exerted a prominent anti-GBM effect in vivo with good safety. Thus, S670, a novel amide derivative of AKBA, could be a potential drug candidate for GBM.
这里,总共设计并合成了 23 种新型 AKBA 酰胺衍生物以增强药理功效,并且发现化合物 S670 的 IC 50远低于 AKBA [ 23 ]。 S670在体外抑制GBM细胞的增殖。以下机制研究表明,S670 通过刺激 ROS 生成诱导 GBM 细胞铁死亡而发挥抗 GBM 活性。此外,S670可以通过ROS激活的Nrf2核转位诱导自噬体生物发生,并通过ROS激活的TFEB核转位刺激溶酶体生物发生。然而,S670通过抑制GBM细胞中STX17的表达,中断自噬体和溶酶体的融合,从而加剧铁死亡。此外,S670在体内发挥了显着的抗GBM作用,且安全性良好。因此,S670,一种新型的 AKBA 酰胺衍生物,可能是治疗 GBM 的潜在候选药物。
Materials and methods 材料和方法
Reagents and antibodies 试剂和抗体
S670 was synthesized and kindly provided by Prof. Jian-you Shi (University of Electronic Science and Technology of China). Reagents and antibodies mentioned in experiments were listed in Supplementary Tables S1 and S2.
S670由电子科技大学史建友教授合成并友情提供。实验中提到的试剂和抗体列于补充表S1和S2中。
Cell culture and proliferation assay
细胞培养和增殖测定
The human GBM cell lines U87 and U251 (American Type Culture Collection, ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, CA, USA) with 10% fetal bovine serum (164210-50, FBS, Procell, China) in a humidified 5% CO2 incubator at 37 °C. CCK-8 (C0039, Beyotime, Shanghai, China) was used for cell viability assay. Cells (3 × 103) were put into 96-well plate per well. After designed treatment, FBS-free DMEM containing 10% CCK-8 reagent was added and A450 nm was measured by FlexStation 3 (Molecular Devices, Sunnyvale, CA, USA) after being incubated at 37 °C.
人 GBM 细胞系 U87 和 U251(美国典型培养物保藏中心,ATCC)在含有 10% 胎牛血清(164210-50,FBS,Procell,中国)的 Dulbecco 改良 Eagle 培养基(DMEM,Gibco,CA,USA)中培养。 37 °C 湿润的 5% CO 2培养箱。 CCK-8(C0039,Beyotime,上海,中国)用于细胞活力测定。每孔将细胞(3×10 3 )放入96孔板中。经过设计处理后,加入含有 10% CCK-8 试剂的不含 FBS 的 DMEM,并在 37 °C 下孵育后,通过 FlexStation 3 (Molecular Devices, Sunnyvale, CA, USA) 测量450 nm 。
DNA synthesis assay DNA合成测定
EdU Kit (C10310, RiboBio, Guangzhou, China) was used for DNA synthesis assay according to the manufacture’s protocol. After being treated with S670, cells were incubated with EdU (50 μM). After being fixed and permeabilized, cells were stained with Apollo 567 and then counterstained with Hoechst 33342. Representative images were captured using a microscope (Nikon, Tokyo, Japan).
EdU 试剂盒(C10310,RiboBio,广州,中国)根据制造商的方案用于 DNA 合成测定。用 S670 处理后,将细胞与 EdU (50 μM) 一起孵育。固定和透化后,细胞用 Apollo 567 染色,然后用 Hoechst 33342 复染。使用显微镜(尼康,东京,日本)捕获代表性图像。
Transwell assay Transwell实验
Transwell Permeable Supports (3342, Corning Costar, Kennebunk, USA) was used according to the manufacture’s protocol. For invasion assay, Matrigel (354234, Corning Costar, Kennebunk, USA) was coated on the bottom of the Transwell insert with 1:7 dilution. Cell suspension (5 × 104 cells/mL, 200 μL) with FBS-free DMEM was seeded in the upper chamber. DMEM with 20% FBS was filled into the lower chamber. After being incubated for 24 h, cells that passed through the polycarbonate membrane was fixed and stained by 1% crystal violet (G1062, Solarbio, Beijing, China). Cells were captured and counted under a microscope. For migration assay, the steps were the same but no Matrigel was coated.
根据制造商的方案使用 Transwell 可渗透支持物(3342,Corning Costar,肯纳邦克,美国)。对于侵袭测定,Matrigel (354234, Corning Costar, Kennebunk, USA) 以 1:7 稀释液涂在 Transwell 插入件的底部。将含有不含 FBS DMEM 的细胞悬液(5 × 10 4 个细胞/mL,200 μL)接种到上室中。将含有 20% FBS 的 DMEM 填充到下室中。孵育24小时后,将穿过聚碳酸酯膜的细胞固定并用1%结晶紫(G1062,Solarbio,北京,中国)染色。捕获细胞并在显微镜下计数。对于迁移测定,步骤相同,但不包被基质胶。
Soft agar colony formation assay
软琼脂集落形成测定
After being treated with S670 at 0, 4, 8, 12 μM for 24 h, about 1 × 103 cells were mixed with 0.35% soft agar (diluted with 20% FBS DMEM), put into six‐well plates and cultured for 2 weeks. The clones were stained with 5 mg/mL MTT, photographed and clone number was counted.
0、4、8、12 μM S670处理24 h后,约1×10 3 个细胞与0.35%软琼脂(用20% FBS DMEM稀释)混合,放入六孔板中培养2几周。将克隆用5 mg/mL MTT染色,拍照并计数克隆数。
Cell cycle and apoptosis assay
细胞周期和细胞凋亡测定
Cells were harvested, fixed and resuspended in propidium iodide (PI) solution (C0080, Solarbio, Beijing, China) or stained with Annexin V and PI Kit (A211-02, Vazyme, Nanjing, China). Cell cycles or cell apoptosis were checked by BD FACSVerse Cytometer (BD, San Diego, CA, USA). The distributions of cell cycle or cell apoptosis rate were analyzed by Flow Jo_V10.
收获细胞,固定并重悬于碘化丙啶 (PI) 溶液(C0080,Solarbio,北京,中国)中或用膜联蛋白 V 和 PI 试剂盒(A211-02,Vazyme,南京,中国)染色。通过 BD FACSVerse 细胞仪(BD,圣地亚哥,加利福尼亚州,美国)检查细胞周期或细胞凋亡。 Flow Jo_V10分析细胞周期或细胞凋亡率的分布。
Western blotting 蛋白质印迹法
Cells were harvested and lysed in RIPA lysis buffer (C1053-100, Gene-Protein Link, Beijing, China) on ice. After being centrifuged, the supernatant was added with SDS-PAGE Loading Buffer (CW0027, CWBIO, Beijing, China) and equivalent amount of 20 μg proteins was separated by SDS-PAGE electrophoresis, transferred to polyvinylidene fluoride (PVDF) membranes (IPVH00010, Millipore, Darmstadt, Germany), sealed up in 5% skim milk (P1622, Applygen, Beijing, China), and then incubated with corresponding diluted primary antibodies at 4 °C overnight. Finally, the membranes were incubated with secondary antibodies. Band intensity was visualized with SuperECL (P1050, Applygen, Beijing, China).
收获细胞并在冰上用 RIPA 裂解缓冲液(C1053-100,Gene-Protein Link,北京,中国)裂解。离心后,上清液中加入 SDS-PAGE Loading Buffer(CW0027,CWBIO,北京,中国),通过 SDS-PAGE 电泳分离等量的 20 μg 蛋白,转移至聚偏二氟乙烯(PVDF)膜(IPVH00010,Millipore) ,达姆施塔特,德国),密封在 5% 脱脂牛奶中(P1622,Applygen,北京,中国),然后与相应稀释的一抗在4℃下孵育过夜。最后,将膜与二抗一起孵育。使用 SuperECL(P1050,Applygen,北京,中国)可视化带强度。
RNA extraction and RT-qPCR
RNA提取和RT-qPCR
Total RNA was extracted using Total RNA Extraction Kit (G01E01S, Gene-Protein Link, Beijing, China). Complementary DNA (cDNA) was reverse-transcribed from total RNA by using SuperRT cDNA Synthesis Kit (CW0741M, CWBIO, Beijing, China). The qPCR analysis was performed with mixture using MagicSYBR Mixture (CW3008M, CWBIO, Beijing, China), RNase-free water (R1600, Solarbio, Beijing, China), and primers (synthesized by Beijing Tsingke Biology Co., Ltd, Supplementary Table S3) on a CFX thermocycler (Bio-Rad, Hercules, CA, USA).
使用总RNA提取试剂盒(G01E01S,Gene-Protein Link,北京,中国)提取总RNA。使用SuperRT cDNA合成试剂盒(CW0741M,CWBIO,北京,中国)从总RNA反转录互补DNA(cDNA)。使用MagicSYBR Mixture(CW3008M,CWBIO,北京,中国)、无RNase水(R1600,Solarbio,北京,中国)和引物(由北京清科生物有限公司合成,补充表S3)进行qPCR分析)在 CFX 热循环仪(Bio-Rad,Hercules,CA,USA)上。
Detection of cell ferrous iron, ROS and lipid ROS
细胞二价铁、ROS和脂质ROS的检测
Cells were harvested after the treatment of S670. The level of Fe2+ was detected using cell ferrous iron colorimetric assay kit (E-BC-K881-M, Elabscience, Wuhan, China). For the detection of ROS and lipid ROS, cells were stained with DCFH-DA (35845, Sigma-Aldrich, Germany) or BODIPY 581/591 C11 (HY-D1301, MedChemExpress, Shanghai, China). After being washed, cells were harvested and the level of reactive oxygen species (ROS) or lipid ROS was imaged under a microscope or detected by flow cytometry.
S670处理后收获细胞。使用细胞亚铁比色测定试剂盒(E-BC-K881-M,Elabscience,武汉,中国)检测Fe 2+水平。为了检测ROS和脂质ROS,细胞用DCFH-DA(35845,Sigma-Aldrich,德国)或BODIPY 581/591 C11(HY-D1301,MedChemExpress,上海,中国)染色。洗涤后,收获细胞,并在显微镜下成像或通过流式细胞仪检测活性氧(ROS)或脂质ROS的水平。
Immunofluorescence 免疫荧光
After designed treatment, cells were fixed, permeabilized, and blocked, then cells were incubated with primary antibodies at recommended dilution at 4 °C overnight. After being washed with PBS, cells were incubated with secondary antibodies, stained with DAPI (C0065, Solarbio, Beijing, China) and then observed under a laser confocal microscope (Leica TCS SP8X, Leica, Germany).
经过设计的处理后,对细胞进行固定、透化和封闭,然后将细胞与建议稀释度的一抗在 4 °C 下孵育过夜。用PBS洗涤细胞后,用二抗孵育,用DAPI(C0065,Solarbio,北京,中国)染色,然后在激光共聚焦显微镜(Leica TCS SP8X,Leica,德国)下观察。
Lysotracker RED staining Lysotracker RED 染色
Cells were stained with LysoRed Tracker (C1046, Beyotime, Shanghai, China) for 10 min at 37 °C. After being washed, the lysosomal mass of cells was analyzed by flow cytometry.
细胞用 LysoRed Tracker(C1046,Beyotime,上海,中国)在 37°C 染色 10 分钟。洗涤后,通过流式细胞术分析细胞的溶酶体团。
Neutral red staining 中性红染色
Neutral Red Staining Solution (C0123, Beyotime, Shanghai, China) was used to analyze the lysosome pH and the stained lysosomes (red) were observed under a microscopy.
使用中性红染色液(C0123,Beyotime,上海,中国)分析溶酶体 pH 值,并在显微镜下观察染色的溶酶体(红色)。
Autophagic flux analysis 自噬通量分析
Briefly, 5 × 104 of cells were seeded and infected with tandem mRFP-GFP-LC3 adenovirus (1 × 108 transducing units/mL, HANBIO, Shanghai, China) in MOI = 1. After being infected for 24 h, autophagosome (yellow puncta) and autolysosome (red puncta) in cells were observed using laser confocal microscopy.
简而言之,接种 5 × 10 4个细胞,并以 MOI = 1 感染串联 mRFP-GFP-LC3 腺病毒(1 × 10 8转导单位/mL,汉宝生物,上海,中国)。感染 24 小时后,自噬体(使用激光共聚焦显微镜观察细胞中的黄色斑点)和自溶酶体(红色斑点)。
RNA interference RNA干扰
The scrambled siRNAs were synthesized by Beijing Tsingke Biology Co., Ltd. and the sequences were listed in Supplementary Table S4. siRNA transfection was performed at approximately 50% confluency using Lipofectamine 3000 reagent (L3000015, Invitrogen, CA, USA). The efficacy of knockdown was verified by Western blotting after transfection for 48 h.
打乱的siRNA由北京清科生物有限公司合成,序列列于补充表S4中。使用 Lipofectamine 3000 试剂(L3000015,Invitrogen,CA,USA)在约 50% 汇合时进行 siRNA 转染。转染48小时后通过Western blotting验证敲低效果。
Co-Immunoprecipitation (Co-IP)
免疫共沉淀 (Co-IP)
Cells were collected and lysed in IP lysis buffer (C1054, Applygen, Beijing, China) on ice. After being centrifugated, the supernatant was incubated with 25 μg/mL of antibodies against LC3 or LAMP2 at 4 °C overnight. After pulling down of immunoprecipitated protein using Protein A + G magnetic beads (P2108, Beyotime, Shanghai, China), interaction between two proteins was analyzed by Western blotting.
收集细胞并在冰上用 IP 裂解缓冲液(C1054,Applygen,北京,中国)裂解。离心后,将上清液与 25 μg/mL LC3 或 LAMP2 抗体在 4 °C 下孵育过夜。使用 Protein A + G 磁珠(P2108,Beyotime,上海,中国)下拉免疫沉淀蛋白后,通过蛋白质印迹分析两种蛋白之间的相互作用。
Transmission electron microscopy (TEM) analysis
透射电子显微镜 (TEM) 分析
Cells were fixed in electron microscope fixative and postfixed with 1% OsO4. The samples were then dehydrated, infiltrated and embedded by epoxy resin. The ultra-structures of mitochondrial and autophagosome were captured by a TEM (HITACHI, Tokyo, Japan).
将细胞固定在电子显微镜固定液中并用1% OsO 4进行后固定。然后将样品脱水、用环氧树脂渗透和包埋。线粒体和自噬体的超微结构由 TEM(HITACHI,东京,日本)捕获。
Xenograft model of nude mice
裸鼠异种移植模型
All animal experiments were conducted in accordance with National Research Council’s Guide for the Care and Use of Laboratory Animals and approved by Institute of Materia Medica, Chinese Academy of Medical Science and Peking Union Medical College (Beijing, China) (No. 00008611). To construct xenograft tumor model, 1 × 106 U87 cells were subcutaneously implanted into the right flanks of the BALB/c nude mice (female, 17–19 g, Charles River, Beijing, China). The mice were randomly divided into four groups (n = 6) when the average tumor volume was more than 100 mm3, and mice received the following treatments: DMSO, S670 (25 mg/kg), S670 (50 mg/kg) and TMZ (10 mg/kg) once a day by irrigation. The tumor volume (V, mm3) was calculated according to V = 0.5 × l × w2 (l = length, w = width in mm). The mice were sacrificed when the largest tumor volume reached 1000 mm3. Tumor tissues were collected, weighed and photographed. Heart, liver, spleen, kidney, lung and brain of mice were collected and weighed to calculate organ index.
所有动物实验均按照国家研究委员会实验动物护理和使用指南进行,并经中国医学科学院药物研究所和北京协和医学院(中国北京)批准(编号:00008611)。为了构建异种移植肿瘤模型,将 1 × 10 6 U87 细胞皮下植入 BALB/c 裸鼠(雌性,17-19 g,Charles River,北京,中国)的右侧腹。当平均肿瘤体积大于100 mm 3时,将小鼠随机分为4组( n =6),小鼠接受以下治疗:DMSO、S670(25 mg/kg)、S670(50 mg/kg)和TMZ (10 mg/kg) 每天一次,通过冲洗。根据V=0.5×l×w 2 (l=长度,w=宽度,单位mm)计算肿瘤体积(V,mm 3 )。当最大肿瘤体积达到1000mm 3时处死小鼠。收集肿瘤组织、称重并拍照。采集小鼠心、肝、脾、肾、肺、脑称重,计算脏器指数。
Hematoxylin-eosin (H&E) staining and immunohistochemistry (IHC)
苏木精-伊红 (H&E) 染色和免疫组织化学 (IHC)
The tumor in vehicle group and S670 (50 mg/kg) group were sectioned, dried, deparaffinized, rehydrated, antigen repaired, incubated with primary antibodies and visualized using diaminobenzidine (DAB) to analyze the expression of Ki67, LC3 and LAMP2. The collected organs in vehicle group and S670 (50 mg/kg) group were sectioned, deparaffinized, rehydrated, and stained with H&E. The representative images were captured under a microscopy.
将媒介物组和S670(50 mg/kg)组的肿瘤切片、干燥、脱蜡、复水、抗原修复、与一抗孵育,并使用二氨基联苯胺(DAB)显色来分析Ki67、LC3和LAMP2的表达。将媒介物组和S670(50mg/kg)组收集的器官切片、脱蜡、复水并用H&E染色。代表性图像是在显微镜下捕获的。
Statistical analysis 统计分析
The results are shown as mean ± standard deviation (SD). Unpaired Student’s t test was used to calculate statistical significance between two groups. One-Way-ANOVA was used to calculate statistical significance between multiple groups. GraphPad Prism 8.0 (GraphPad Software Inc., San Diego, CA, USA) was used to perform statistical significance. It was considered statistically significant when P < 0.05.
结果显示为平均值±标准差(SD)。未配对的学生t检验用于计算两组之间的统计显着性。使用单因素方差分析来计算多组之间的统计显着性。 GraphPad Prism 8.0(GraphPad Software Inc.,圣地亚哥,加利福尼亚州,美国)用于执行统计显着性分析。当P <0.05时被认为具有统计学意义。
Results 结果
S670 inhibited the proliferation, migration, invasion and colony formation of GBM cells
S670抑制GBM细胞的增殖、迁移、侵袭和集落形成
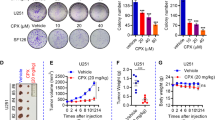
Based on our previous study [23, 24], AKBA could induce apoptosis, block cell cycle, and ameliorate aberrant metabolic landscape in GBM with an IC50 of about 30 μM at 24 h. To enhance pharmacological efficacy and improve oral bioavailability, a series of 23 amide derivatives of S670 were synthesized and screened in vitro for the anti-proliferation effect on GBM cells, which has been applied for China invention patent (No. 202111625269.5). Among them, S670 exhibited optimal anti-proliferation effect and was chosen for further validation. The process of design, synthesis, drug screening and structure of S670 were shown in Fig. 1a.
根据我们之前的研究 [ 23 , 24 ],AKBA 可以诱导细胞凋亡、阻断细胞周期并改善 GBM 中的异常代谢景观,24 小时时的 IC 50约为 30 μM。为了增强药理功效,提高口服生物利用度,合成了一系列23种S670酰胺衍生物,并在体外筛选了其对GBM细胞的抗增殖作用,已申请中国发明专利(专利号:202111625269.5)。其中,S670表现出最佳的抗增殖效果,被选择进行进一步验证。 S670的设计、合成、药物筛选和结构流程如图1a所示。
图1:S670在体外抑制GBM细胞的增殖。

a The flow chart of evaluation of the anti-GBM activity of the novel synthesized amide AKBA derivatives in vitro. b Cell viability was determined by CCK-8 assay after incubation with 0, 0.5, 1, 2, 4, 8 and 16 μM S670 for 24, 48 and 72 h. IC50 at 24 h, 48 h and 72 h was calculated and noted, respectively. c The DNA synthesis rate was measured by DNA-EdU assay after treatment of 0, 2, 4 and 8 μM S670 after 24 h. Scale bar = 100 μm. d The colony formation ability was evaluated after treatment of 0, 4, 8 and 12 μM S670. e Expression of E-Cadherin and N-Cadherin was detected by Western blotting after treatment of 0, 4, 8 and 12 μM S670. f Flow cytometry using PI staining was used to analyze the cell cycle phase distribution after treatment of 0, 4, 8 and 12 μM S670. The data were performed in triplicate and shown as the mean ± SD.
a新型合成的酰胺AKBA衍生物的体外抗GBM活性评价流程图。 b用 0、0.5、1、2、4、8 和 16 μM S670 孵育 24、48 和 72 小时后,通过 CCK-8 测定法测定细胞活力。分别计算并记录24小时、48小时和72小时的IC 50 。 c用 0、2、4 和 8 μM S670 处理 24 小时后,通过 DNA-EdU 测定测量 DNA 合成率。比例尺 = 100 μm。 d 0、4、8和12μM S670处理后评估集落形成能力。 e用0、4、8和12 μM S670处理后,通过Western blotting检测E-Cadherin和N-Cadherin的表达。 f使用 PI 染色的流式细胞术分析 0、4、8 和 12 μM S670 处理后的细胞周期时相分布。数据一式三份进行并显示为平均值±SD。
To evaluate the anti-GBM effect of S670 in vitro, a series of assays were carried out. CCK-8 assay indicated that S670 inhibited the growth of GBM cells dose and time-dependently. The IC50 of S670 at 24 h, 48 h, and 72 h were 5.961 μM, 5.738 μM, and 4.448 μM on U87 cells. The IC50 of S670 at 24 h, 48 h, and 72 h on U251 cells were 8.023 μM, 5.119 μM, and 4.875 μM, respectively (Fig. 1b). Compared with AKBA, S670 exhibited much stronger anti-proliferation activity on GBM cells [23]. EdU-DNA synthesis assay indicated that S670 inhibited DNA synthesis of GBM cells dose-dependently in 24 h and 48 h (Fig. 1c, Supplementary Fig. S1a). Moreover, S670 significantly inhibited the ability of migration and invasion (Supplementary Fig. S1b–d) as well as colony formation (Fig. 1d, Supplementary Fig. S1e) dose-dependently. Loss of E-cadherin and overexpression of N-cadherin are involved in tumor progression and metastasis [25, 26]. As shown in Fig. 1e, the expression of E-cadherin was increased but N-cadherin was decreased after the treatment of S670. Additionally, PI staining detected by flow cytometry showed that cell cycle was blocked at the G2/M phase dose-dependently by S670 (Fig. 1f, Supplementary Fig. S1f). All in all, S670 exhibited prominent anti-proliferation effect of GBM cells in vitro.
为了评估 S670 的体外抗 GBM 作用,进行了一系列测定。 CCK-8实验表明S670对GBM细胞生长的抑制作用呈剂量和时间依赖性。 S670 在 U87 细胞上 24 h、48 h 和 72 h 时的 IC 50分别为 5.961 μM、5.738 μM 和 4.448 μM。 S670在24小时、48小时和72小时对U251细胞的IC 50分别为8.023μM、5.119μM和4.875μM(图1b )。与AKBA相比,S670对GBM细胞表现出更强的抗增殖活性[ 23 ]。 EdU-DNA合成测定表明,S670在24小时和48小时内剂量依赖性地抑制GBM细胞的DNA合成(图1c ,补充图S1a )。此外,S670 显着抑制迁移和侵袭的能力(补充图S1b-d )以及集落形成(图1d ,补充图S1e ),且呈剂量依赖性。 E-钙粘蛋白的缺失和N-钙粘蛋白的过度表达与肿瘤进展和转移有关[ 25 , 26 ]。如图1e所示,S670处理后E-钙粘蛋白的表达增加,但N-钙粘蛋白的表达减少。此外,流式细胞术检测到的PI染色显示,细胞周期被S670剂量依赖性地阻断在G 2 /M期(图1f ,补充图S1f )。总而言之,S670在体外表现出显着的抗GBM细胞增殖作用。
S670 stimulated ROS generation and induced ferroptosis in GBM cells
S670 刺激 GBM 细胞中 ROS 的产生并诱导铁死亡
The mechanisms of the anti-GBM activity of S670 were further explored. As previously reported, AKBA could induce apoptosis of GBM cells [23]. To explore whether S670 could also induce apoptosis in GBM cells, flow cytometry analysis was performed after the treatment of 0, 4, 8, 12 μM S670. Results showed that S670 significantly increased cell death (Supplementary Fig. S2a). However, the expression of proteins associated with apoptosis including Caspase 3, Cleaved-caspase 3 and Bcl-2 was not significantly changed after being treated with S670 for 0 h, 1 h, 3 h, 6 h, 12 h and 24 h (Supplementary Fig. S2b). Moreover, cell death induced by S670 could also not be rescued by Z-VAD-FMK or Necrostatin-1 (Nec-1) (Supplementary Fig. S2c, d). All these results suggested that apoptosis or necroptosis might not be the major contributor to GBM cell death caused by S670.
进一步探讨了S670的抗GBM活性机制。如先前报道,AKBA可以诱导GBM细胞凋亡[ 23 ]。为了探讨S670是否也能诱导GBM细胞凋亡,在0、4、8、12 μM S670处理后进行流式细胞术分析。结果显示,S670 显着增加细胞死亡(补充图S2a )。然而,S670处理0小时、1小时、3小时、6小时、12小时和24小时后,与细胞凋亡相关的蛋白(包括Caspase 3、Cleaved-caspase 3和Bcl-2)的表达没有显着变化(补充图S2b )。此外,Z-VAD-FMK 或 Necrostatin-1 (Nec-1) 也无法挽救 S670 诱导的细胞死亡(补充图S2 c、d)。所有这些结果表明细胞凋亡或坏死性凋亡可能不是 S670 引起的 GBM 细胞死亡的主要因素。
AKBA has been reported to possess potent anti-inflammatory activity [27]. To explore whether S670 also possessed anti-inflammatory activity in GBM cells, ROS level was checked after the treatment of S670. However, intracellular ROS level was significantly increased by S670, which was monitored under the fluorescence microscope (Fig. 2a) or by flow cytometry (Fig. 2b) after dying with DCFH-DA. And ROS scavenger N-acetylcysteine (NAC) could eliminate the ROS generation in GBM cells and attenuated S670-induced cell death (Fig. 2c, d). Mitochondria is the main source of ROS generation in cancer cells [28]. To determine the source of ROS generation, mtSOX Deep Red was used for measurement of mitochondrial ROS. And results showed that S670 induced the generation of mitochondrial ROS (Supplementary Fig. S3a). The above results demonstrated that S670-induced mitochondrial ROS generation mainly contributed to S670-induced cell death.
据报道 AKBA 具有有效的抗炎活性 [ 27 ]。为了探讨S670是否也具有GBM细胞的抗炎活性,在S670处理后检查ROS水平。然而,S670 显着增加了细胞内 ROS 水平,这是在用 DCFH-DA 染色后在荧光显微镜下(图2a )或通过流式细胞术(图2b )监测的。 ROS清除剂N-乙酰半胱氨酸(NAC)可以消除GBM细胞中ROS的产生并减弱S670诱导的细胞死亡(图2c,d )。线粒体是癌细胞中ROS产生的主要来源[ 28 ]。为了确定 ROS 产生的来源,使用 mtSOX Deep Red 测量线粒体 ROS。结果表明,S670 诱导线粒体 ROS 的产生(补充图S3a )。上述结果表明,S670诱导的线粒体ROS生成主要是S670诱导的细胞死亡的原因。
图 2:S670 刺激 GBM 细胞中 ROS 的产生并诱导铁死亡。

a ROS level was measured after being stained with DCFH-DA and captured by a fluorescence microscope after treated by S670 (6 μM) for 24 h. The nucleus was stained by DAPI. Scale bar = 100 μm. b Relative ROS level was detected by flow cytometry after being stained with DCFH-DA. c CCK8 assay was carried out to explore the effect of NAC (5 mM) on cell death caused by S670 (6 μM) after treatment for 24 h. d The cell death rate was detected by flow cytometry and corresponding quantification results were shown. e CCK8 assay was performed to explore the effect of Fer-1 (1 μM) on cell viability loss caused by S670 (6 μM) after treatment for 24 h. f Relative lipid ROS level in U87 and U251 cells was detected by flow cytometry in each group after being stained with C11 BODIPY 581/591 and corresponding quantification results were shown. g The representative images of mitochondrial captured by TEM after 6 μM S670 treatment for 24 h. Scale bar = 1 μm. h Expression of COX-2 and GPX4 in GBM cells was detected by Western blotting after treatment with 0, 4, 8 and 12 μM S670. The data were performed in triplicate and shown as the mean ± SD. **P < 0.01, ****P < 0.0001 compared to NC group. #P < 0.05, ##P < 0.01, ###P < 0.001, ####P < 0.0001 compared to S670 treatment group.
S670 (6 μM) 处理 24 h 后,用 DCFH-DA 染色并通过荧光显微镜捕获后测量 ROS水平。细胞核用 DAPI 染色。比例尺 = 100 μm。 b DCFH-DA染色后通过流式细胞术检测相对ROS水平。 c进行CCK8测定以探讨NAC(5 mM)对S670(6 μM)处理24小时后引起的细胞死亡的影响。 d流式细胞术检测细胞死亡率并给出相应的定量结果。 e CCK8 检测旨在探讨 Fer-1 (1 μM) 对 S670 (6 μM) 处理 24 小时后引起的细胞活力丧失的影响。 f C11 BODIPY 581/591染色后,采用流式细胞仪检测各组U87和U251细胞中相对脂质ROS水平,并给出相应的定量结果。 g 6 μM S670 处理 24 小时后通过 TEM 捕获的线粒体的代表性图像。比例尺 = 1 μm。 h用0、4、8和12 μM S670处理后,通过Western blotting检测GBM细胞中COX-2和GPX4的表达。数据一式三份进行并显示为平均值±SD。 ** 与 NC 组相比, P < 0.01,**** P < 0.0001。与 S670 治疗组相比, # P < 0.05, ## P < 0.01, ### P < 0.001, #### P < 0.0001。
ROS induced lipid peroxidation and played a critical role in ferroptosis [29]. Moreover, the generation of mitochondrial ROS is critical for lipid peroxidation and ferroptosis [30]. To verify whether S670 could induce ferroptosis in GBM cells, the ferroptosis inhibitor ferrostatin-1 (Fer-1) was used to check S670-induced cell death. It was shown that Fer-1 could significantly attenuate S670-induced cell death (Fig. 2e). To further verify that S670 could induce ferroptosis in GBM cells, the level of cell ferrous iron, lipid ROS level and the ultra-structure of mitochondrial were evaluated after the treatment of S670. Ferrous iron was measured using colorimetric assay and the level of Fe2+ was significantly upregulated in U87 and U251 cells (Supplementary Fig. S3b), accompanied with the decreasing level of FTH (Ferritin heavy chain) (Supplementary Fig. S3c). Flow cytometry analysis showed that S670 could significantly induce lipid peroxidation in GBM cells (Fig. 2f). Moreover, TEM was used to observe the ultrastructure of mitochondria and results showed that mitochondria in U87 and U251 cells were shrunken with an increase in mitochondrial membrane density and decrease of mitochondrial cristae after the treatment of S670, which is one of the markers of ferroptosis [5] (Fig. 2g). Activation of COX2 (PTGS2) and impaired GPX4 activity are two critical ferroptosis biomarkers [31]. As shown in Fig. 2h, the expression of COX2 was increased but the expression of GPX4 was inhibited dose-dependently after the treatment of S670, which further demonstrated S670 induced ferroptosis. Altogether, S670 stimulated ROS generation in GBM cells and further induced ferroptosis which led to cell death.
ROS诱导脂质过氧化并在铁死亡中发挥关键作用[ 29 ]。此外,线粒体ROS的产生对于脂质过氧化和铁死亡至关重要[ 30 ]。为了验证S670是否可以诱导GBM细胞铁死亡,使用铁死亡抑制剂ferrostatin-1(Fer-1)来检查S670诱导的细胞死亡。结果显示Fer-1可以显着减弱S670诱导的细胞死亡(图2e )。为了进一步验证S670能够诱导GBM细胞铁死亡,检测S670处理后细胞亚铁水平、脂质ROS水平和线粒体超微结构。使用比色法测量亚铁,U87和U251细胞中Fe 2+的水平显着上调(补充图S3b ),同时伴随着FTH(铁蛋白重链)水平的下降(补充图S3c )。流式细胞术分析表明,S670可以显着诱导GBM细胞中的脂质过氧化(图2f )。此外,利用TEM观察线粒体超微结构,结果显示,S670处理后,U87和U251细胞中线粒体出现萎缩,线粒体膜密度增加,线粒体嵴减少,这是铁死亡的标志之一。 5 ](图2g )。 COX2 (PTGS2) 的激活和 GPX4 活性受损是两个关键的铁死亡生物标志物 [ 31 ]。如图2h所示,S670处理后COX2的表达增加,而GPX4的表达呈剂量依赖性抑制,这进一步证明S670诱导铁死亡。 总而言之,S670 刺激 GBM 细胞中 ROS 的产生,并进一步诱导铁死亡,从而导致细胞死亡。
Since GPX4 plays an important role in the regulation of ferroptosis, we further explored how S670 reduced the expression of GPX4 protein. To investigate whether S670 inhibited the transcription of GPX4, the mRNA level of GPX4 was analyzed by RT-qPCR after the treatment of S670. However, S670 had no effect on the transcription level of GPX4 in U87 and U251 cells (Supplementary Fig. S3d), which was not consistent with the change of protein levels of GPX4. Thus, we speculated S670 might reduce the protein level of GPX4 by increasing protein degradation of GPX4. Since GPX4 has been reported to be degraded via ubiquitination or via the lysosome pathway [32], the proteosome inhibitor MG-132 and lysosome inhibitor Bafilomycin A1 (BafA1) were used to verify the degradation pathway of GPX4 after the treatment of S670. The treatment of BafA1 but not MG-132 could rescue the decreased level of GPX4, which demonstrated that S670 could promote the degradation of GPX4 via the lysosomal pathway (Supplementary Fig. S3e).
由于GPX4在铁死亡的调节中发挥重要作用,我们进一步探讨了S670如何降低GPX4蛋白的表达。为了研究S670是否抑制GPX4的转录,在S670处理后通过RT-qPCR分析GPX4的mRNA水平。然而,S670对U87和U251细胞中GPX4的转录水平没有影响(补充图S3d ),这与GPX4蛋白水平的变化不一致。因此,我们推测S670可能通过增加GPX4的蛋白质降解来降低GPX4的蛋白质水平。由于已有报道GPX4通过泛素化或溶酶体途径降解[ 32 ],因此使用蛋白酶体抑制剂MG-132和溶酶体抑制剂巴弗洛霉素A1(BafA1)来验证S670处理后GPX4的降解途径。 BafA1而非MG-132的处理可以挽救GPX4水平的降低,这表明S670可以通过溶酶体途径促进GPX4的降解(补充图S3e )。
S670 induced protective autophagy via Nrf2 activation
S670 通过 Nrf2 激活诱导保护性自噬
The burst of ROS generation could activate autophagy [33]. To confirm whether S670 affected the autophagy flux in GBM, the expression of LC3 and p62 was checked by Western blotting. The expression of LC3B and p62 was increased dose and time-dependently after the treatment of S670 (Fig. 3a, b). Meanwhile, the transcriptional level of LC3 was also significantly upregulated after the treatment of S670 (Supplementary Fig. S4a). The transcription activation and accumulation of LC3B indicated S670 might activate autophagy in GBM cells. However, the accumulation of p62 indicated that S670 could impair the autophagic degradation [34]. S670 also could induce a significant increase of EGFP-LC3 puncta in U87 and U251 cells, which also reflected the accumulation of autophagosomes (Fig. 3c). Similarly, it was shown in Fig. 3d that increased autophagosomes could be observed by TEM in GBM cells after being treated by S670. To further verify the change of autophagy flux, rapamycin (Rapa) and chloroquine (CQ) were used to examine the impact on LC3B accumulation. As shown in Fig. 3e, the combination of S670 and Rapa or S670 and CQ could further enhance the accumulation of LC3B than using Rapa or CQ singly, which confirmed that S670 might activate autophagy in GBM cells. As autophagy plays double functions in cancer, we further verify whether S670 induced protective autophagy or activated autophagy, which contributed to the anti-tumor effect in GBM cells. Beclin-1 is a critical factor in the regulation of autophagosome formation and maturation [35]. We knocked down Beclin-1 expression with siBeclin-1 in GBM cells and siBeclin-1-2 exhibited good gene silencing efficacy (Supplementary Fig. S4b, c). Results showed that U87 and U251 cells are more sensitive to S670 after knockdown of the expression of Beclin-1 (Fig. 3f). All in all, S670 could induce autophagy in GBM cells, which might be a self-protection mechanism.
ROS 产生的爆发可以激活自噬[ 33 ]。为了确认 S670 是否影响 GBM 中的自噬流,通过蛋白质印迹检查 LC3 和 p62 的表达。 S670处理后LC3B和p62的表达呈剂量和时间依赖性增加(图3a,b )。同时,S670处理后LC3的转录水平也显着上调(补充图S4a )。 LC3B 的转录激活和积累表明 S670 可能激活 GBM 细胞中的自噬。然而,p62 的积累表明 S670 可能会损害自噬降解[ 34 ]。 S670还可以诱导U87和U251细胞中EGFP-LC3斑点的显着增加,这也反映了自噬体的积累(图3c )。同样,图3d显示,经S670处理后,通过TEM可以观察到GBM细胞中自噬体的增加。为了进一步验证自噬通量的变化,使用雷帕霉素(Rapa)和氯喹(CQ)来考察对LC3B积累的影响。如图3e所示,S670与Rapa或S670与CQ的组合比单独使用Rapa或CQ能够进一步增强LC3B的积累,这证实了S670可能激活GBM细胞中的自噬。由于自噬在癌症中发挥双重作用,我们进一步验证S670是否诱导保护性自噬或激活自噬,从而有助于GBM细胞的抗肿瘤作用。 Beclin-1 是调节自噬体形成和成熟的关键因素[ 35 ]。我们用 siBeclin-1 敲低 GBM 细胞中的 Beclin-1 表达,siBeclin-1-2 表现出良好的基因沉默功效(补充图 1)。S4 b、c)。结果显示,敲低Beclin-1的表达后,U87和U251细胞对S670更敏感(图3f )。总而言之,S670可以诱导GBM细胞自噬,这可能是一种自我保护机制。
图 3:S670 通过 Nrf2 激活诱导保护性自噬。

a Expression of LC3B and p62 was checked by Western blotting after treatment with 0, 4, 8 and 12 μM S670. b Expression of LC3B and p62 was checked by Western blotting after treatment with S670 for 0 h, 1 h, 3 h, 6 h, 12 h and 24 h. c Cells stably expressing GFP-LC3 were treated with S670 (6 μM) for 24 h and autophagosomes were observed and counted. d The representative images of autophagic vehicles in GBM cells after treatment of 6 μM S670. Scale bar = 2 μm. e Expression of LC3B of U87 and U251 cells was checked by Western blotting after treatment of rapamycin (Rapa, 500 nM), chloroquine (CQ, 10 μM) or S670 (6 μM) respectively or combination treatment with Rapa plus S670 or CQ plus S670 for 24 h. f CCK8 assay was performed to explore the effect of the ablation of Beclin-1 on cell viability loss caused by S670 (6 μM) after treatment for 24 h. g Western blotting was performed to investigate the effect of NAC (5 mM) on LC3B upregulation caused by S670 (6 μM) after treatment with 24 h. h Expression of Nrf2, Keap1, and HO-1 was checked by Western blotting after treatment of 0, 4, 8 and 12 μM S670 for 24 h. i Expression of Nrf2 and LC3B was detected by Western blotting after treatment of S670 (6 μM), ML385 (10 μM) or combination treatment after 24 h. j U87 and U251 cells were treated with S670 (6 μM), NAC (5 mM) or the combination treatment after 24 h and then cytosolic and nuclear expression of Nrf2 was detected by Western blotting. The data were performed in triplicate and shown as the mean ± SD, *P < 0.05, ****P < 0.0001 compared to the NC group, ####P < 0.0001 compared to S670 treatment group.
a 用 0、4、8 和 12 μM S670 处理后,通过蛋白质印迹检查 LC3B 和 p62 的表达。 b用S670处理0小时、1小时、3小时、6小时、12小时和24小时后,通过蛋白质印迹检查LC3B和p62的表达。 c用S670(6 μM)处理稳定表达GFP-LC3的细胞24 h,观察自噬体并计数。 d 6 μM S670 处理后 GBM 细胞中自噬载体的代表性图像。比例尺 = 2 μm。 e分别用雷帕霉素(Rapa,500 nM)、氯喹(CQ,10 μM)或S670(6 μM)处理或用Rapa加S670或CQ加S670联合处理后,通过Western blotting检查U87和U251细胞LC3B的表达24小时。 f CCK8 检测是为了探讨 Beclin-1 的消融对 S670 (6 μM) 处理 24 小时后引起的细胞活力丧失的影响。 g进行蛋白质印迹以研究 NAC (5 mM) 对 S670 (6 μM) 处理 24 小时后引起的 LC3B 上调的影响。 h用 0、4、8 和 12 μM S670 处理 24 小时后,通过 Western blotting 检查 Nrf2、Keap1 和 HO-1 的表达。 i S670 (6 μM)、ML385 (10 μM) 或联合处理 24 h 后,通过 Western blotting 检测 Nrf2 和 LC3B 的表达。 j U87和U251细胞用S670(6μM)、NAC(5mM)或联合处理24小时后,然后通过Western blotting检测胞浆和核中Nrf2的表达。数据一式三份进行,显示为平均值±SD,* P < 0.05,**** P < 0.0001 与 NC 组相比, #### P < 0.0001 与 S670 治疗组相比。
Autophagy could be activated via the mTOR-dependent pathway, such as PI3K/Akt/mTOR signaling or the mTOR-independent pathway [36]. To further elucidate how S670 activated autophagy, we first explored whether the mTOR-dependent pathway was upstream of S670-stimulated autophagosome biogenesis. However, no obvious alteration in the expression of mTOR and p-mTOR could be observed after the treatment of S670 (Supplementary Fig. S4d). Considering that oxidative stress could promote autophagy via activation of Nrf2 [37], we speculated that ROS generation stimulated by S670 might activate Nrf2 and further promote autophagy in GBM cells. As expected, NAC could alleviate the accumulation of LC3B induced by S670 in U87 and U251 cells (Fig. 3g). And S670 activated Nrf2/Keap1/HO-1 pathway in dose-dependent manner (Fig. 3h). Furthermore, Nrf2-specific inhibitor ML385 could attenuate the activation of Nrf2 and LC3B caused by S670 (Fig. 3i). To confirm whether S670 increased nuclear translocation of Nrf2, Western blotting was carried out to detect the cytosolic and nuclear expression of Nrf2. As shown in Fig. 3j, the expression of Nrf2 in both cytoplasm and nucleus was increased after the treatment of S670 while such an effect could be mitigated by NAC. Collectively, these results demonstrated that S670 activated autophagy via ROS-mediated Nrf2 activation.
自噬可以通过 mTOR 依赖性途径激活,例如 PI3K/Akt/mTOR 信号传导或 mTOR 独立途径 [ 36 ]。为了进一步阐明 S670 如何激活自噬,我们首先探讨 mTOR 依赖性途径是否是 S670 刺激的自噬体生物发生的上游。然而,在S670处理后,没有观察到mTOR和p-mTOR表达的明显变化(补充图S4d )。考虑到氧化应激可以通过激活Nrf2促进自噬[ 37 ],我们推测S670刺激的ROS生成可能会激活Nrf2并进一步促进GBM细胞的自噬。正如所预期的,NAC可以减轻S670在U87和U251细胞中诱导的LC3B的积累(图3g )。并且S670以剂量依赖性方式激活Nrf2/Keap1/HO-1通路(图3h )。此外,Nrf2特异性抑制剂ML385可以减弱S670引起的Nrf2和LC3B的激活(图3i )。为了证实S670是否增加Nrf2的核转位,进行Western blotting检测Nrf2的胞质和核表达。如图3j所示,S670处理后细胞质和细胞核中Nrf2的表达均增加,而NAC可以减轻这种影响。总的来说,这些结果表明 S670 通过 ROS 介导的 Nrf2 激活来激活自噬。
S670 could induce lysosome biogenesis via ROS-stimulated TFEB nuclear translocation
S670 可以通过 ROS 刺激的 TFEB 核易位诱导溶酶体生物发生
Apart from the novel discovery of S670 in the regulation of autophagy, we also observed the dramatic change in morphology of GBM cells after the treatment of S670. Obvious cytoplasmic vacuolization was observed after the treatment of S670 (Supplementary Fig. S5a), which was also reported by Gaurav et al. when cells were treated by 5 WX8-family molecules [38]. The accumulation of cytoplasmic vacuoles was identified as enlarged lysosomes. Lysosomes are the major digestive organelles responsible for the degradation of autophagy substrate [39]. To validate whether the cytoplasmic vacuolization might be the increasing lysosomes induced by S670, the expression of LAMP1 and LAMP2 were detected, which are accounted for about 50% of all proteins of the lysosome membrane [40]. The mRNA levels of LAMP1 and LAMP2 in GBM cells were upregulated after the treatment of S670 (Supplementary Fig. S5b). S670 could also upregulate the protein expression of LAMP2 dose- and time-dependently (Fig. 4a, b). Moreover, S670 significantly increased the lysosome mass in GBM cells, which was monitored by immunofluorescence (Fig. 4c). LysoRed Tracker staining also showed that more lysosomes were stained and detected by flow cytometry (Fig. 4d, Supplementary Fig. S5c). All the above results demonstrated that S670 indeed induced lysosome biogenesis in U87 and U251 cells. Unlike compounds that might affect lysosomal pH [41] or result in lysosomal disorder [42], neutral red staining [43] showed that S670 had no effect on lysosomal pH but BafA1, an H+-ATPase inhibitor, induced lysosomal deacidification and caused that lysosome could not be stained cherry by neutral red (Fig. 4e). Similar results were observed using LysoRed Tracker (Supplementary Fig. S5d). Additionally, S670 did not affect the expression of CTSD (Supplementary Fig. S5e), an essential protease that participated in the regulation of lysosomal proteolytic activity [44]. The above results suggested that S670 induced lysosome biogenesis without affecting the lysosomal function [45].
除了S670在自噬调节方面的新发现外,我们还观察到S670处理后GBM细胞形态发生了巨大的变化。在S670处理后观察到明显的细胞质空泡化(补充图S5a ),这也是Gaurav等人报道的。当细胞被 5 个 WX8 家族分子处理时 [ 38 ]。细胞质空泡的积累被鉴定为扩大的溶酶体。溶酶体是负责自噬底物降解的主要消化细胞器[ 39 ]。为了验证细胞质空泡化是否可能是S670诱导的溶酶体增加,检测了LAMP1和LAMP2的表达,它们约占溶酶体膜所有蛋白质的50%[ 40 ]。 S670处理后GBM细胞中LAMP1和LAMP2的mRNA水平上调(补充图S5b )。 S670还可以剂量和时间依赖性上调LAMP2的蛋白表达(图4a,b )。此外,S670显着增加了GBM细胞中的溶酶体质量,这是通过免疫荧光监测的(图4c )。 LysoRed Tracker染色还显示更多的溶酶体被染色并通过流式细胞术检测到(图4d ,补充图S5c ) 。上述结果均表明S670确实诱导了U87和U251细胞中的溶酶体生物合成。 与可能影响溶酶体pH值[ 41 ]或导致溶酶体紊乱[ 42 ]的化合物不同,中性红染色[ 43 ]表明S670对溶酶体pH值没有影响,但H + -ATP酶抑制剂BafA1诱导溶酶体脱酸并导致溶酶体不能被中性红染色樱桃(图4e )。使用 LysoRed Tracker 观察到类似的结果(补充图S5d ) 。此外,S670不影响CTSD的表达(补充图S5 e ),CTSD是一种参与溶酶体蛋白水解活性调节的重要蛋白酶[ 44 ]。上述结果表明S670诱导溶酶体生物合成而不影响溶酶体功能[ 45 ]。
图 4:S670 通过 ROS 刺激的 TFEB 核转位诱导溶酶体生物发生。

a Expression of LAMP2 was detected by Western blotting after treatment of 0, 4, 8 and 12 μM S670 for 24 h. b Expression of LAMP2 was checked by Western blotting after treatment with S670 for 0 h, 1 h, 3 h, 6 h, 12 h and 24 h. c The immunofluorescence of LAMP2 was observed after the treatment of S670 (6 μM). Nuclei were stained with DAPI (blue). d After being treated with 6 μM S670 for 24 h, cells were stained with LysoRed Tracker and the lysosomal mass was determined by flow cytometry. e Cells were singly treated with S670 (6 μM) or Baf A1 (100 nM) or in combination for 24 h and then stained with Neutral Red. The representative images were shown. Scale bar = 50 μm. f Cells were singly treated with S670 (6 μM) or NAC (5 mM) or cotreated with them for 24 h. The expression of LAMP2 was determined by Western blotting. g Effects of S670 (6 μM, 24 h), NAC (5 mM), or cotreatment of S670 and NAC on TFEB nuclear translocation in GBM cells were detected by an anti-human TFEB (green) antibody and observed by confocal microscopy. Nuclei were counterstained with DAPI (blue). h Cells were treated with S670 (6 μM) or cotreated with S670 and NAC (5 mM) for 24 h. The distribution of TFEB in cytoplasm and nuclei fractions was determined by Western blotting. i After transfection of three TFEB siRNAs and control siRNA for 72 h, the expression of LAMP2 in U87 cells was detected by Western blotting. j U87 cells transfected with si-TFEB or siNC were treated with S670 (6 μM) for 24 h and the expression of LAMP2 was detected by Western blotting. The data were performed in triplicate and shown as the mean ± SD. *P < 0.05, ***P < 0.001 compared to the NC group.
a 0、4、8和12 μM S670处理24 h后,通过Western blotting检测LAMP2的表达。 b用S670处理0小时、1小时、3小时、6小时、12小时和24小时后,通过Western印迹检查LAMP2的表达。 c S670 (6 μM)处理后观察LAMP2的免疫荧光。细胞核用 DAPI(蓝色)染色。 d用 6 μM S670 处理 24 h 后,用 LysoRed Tracker 对细胞进行染色,并通过流式细胞术测定溶酶体质量。 e细胞用 S670 (6 μM) 或 Baf A1 (100 nM) 单独处理或组合处理 24 小时,然后用中性红染色。显示了代表性图像。比例尺 = 50 μm。 f细胞用 S670 (6 μM) 或 NAC (5 mM) 单独处理或与它们共同处理 24 小时。通过蛋白质印迹法测定LAMP2的表达。 g通过抗人 TFEB(绿色)抗体检测 S670(6 μM,24 小时)、NAC(5 mM)或 S670 和 NAC 共处理对 GBM 细胞中 TFEB 核转位的影响,并通过共聚焦显微镜观察。细胞核用 DAPI(蓝色)复染。 h将细胞用 S670 (6 μM) 处理或用 S670 和 NAC (5 mM) 共处理 24 小时。通过蛋白质印迹法确定 TFEB 在细胞质和细胞核部分中的分布。 i转染3个TFEB siRNA和对照siRNA 72 h后,Western blotting检测U87细胞中LAMP2的表达。 j转染si-TFEB或siNC的U87细胞用S670(6 μM)处理24 h,Western blotting检测LAMP2的表达。数据一式三份进行并显示为平均值±SD。 * P < 0.05,*** P < 0。001与NC组相比。
It had been reported that ROS could induce TFEB nuclear translocation and regulate lysosome biogenesis [46]. Moreover, NAC, a ROS scavenger, could attenuate S670-induced LAMP2 upregulation (Fig. 4f). To demonstrate whether S670 promoted lysosome biogenesis via ROS stimulated TFEB nuclear translocation, the distribution of TFEB in cytosol and nucleus was detected after the treatment of S670. It was shown that S670 induced TFEB nuclear translocation and ROS scavenger NAC could attenuate such an accumulation, which was confirmed by immunofluorescence and Western blotting (Fig. 4g, h). To further confirm the regulation of TFEB on lysosome biogenesis, TFEB-specific siRNAs were used for further validation. As expected, knocking down TFEB could also mitigate the upregulation of LAMP2 induced by S670 (Fig. 4i, j) and sensitize U87 cells to S670 (Supplementary Fig. S5f, g). Together, these results demonstrated that S670 induced lysosome biogenesis via ROS-stimulated TFEB nuclear translocation without altering lysosome biological functions.
据报道,ROS可以诱导TFEB核转位并调节溶酶体生物发生[ 46 ]。此外,NAC(一种ROS清除剂)可以减弱S670诱导的LAMP2上调(图4f )。为了证明S670是否通过ROS刺激TFEB核转位促进溶酶体生物合成,在S670处理后检测了TFEB在细胞质和细胞核中的分布。结果表明,S670 诱导的 TFEB 核易位和 ROS 清除剂 NAC 可以减弱这种积累,这通过免疫荧光和蛋白质印迹得到证实(图4g,h )。为了进一步确认 TFEB 对溶酶体生物发生的调节,使用 TFEB 特异性 siRNA 进行进一步验证。正如预期的那样,敲低 TFEB 还可以减轻 S670 诱导的 LAMP2 上调(图4i,j )并使 U87 细胞对 S670 敏感(补充图S5f,g )。总之,这些结果表明,S670 通过 ROS 刺激的 TFEB 核易位诱导溶酶体生物发生,而不改变溶酶体生物学功能。
S670 impaired the autophagosome-lysosome fusion by inhibiting the expression of STX17
S670通过抑制STX17的表达来损害自噬体-溶酶体融合
Although S670 could initiate autophagy and induce the biogenesis of lysosomes, the elevated level of p62 indicated that S670 might impair autophagy degradation [47]. To examine whether S670 affected late-stage autophagy, autophagy flux was checked after the treatment of S670. First, U87 and U251 cells stably expressing mRFP-GFP-tagged LC3B were further treated with S670 for further confirmation of autophagy flux. Results suggested that autophagy flux was blocked after the treatment of S670, as increased yellow dots (autophagosomes) and decreased red dots (autolysosomes) observed in U87 and U251 cells (Fig. 5a, Supplementary Fig. S6a). Moreover, S670 treatment contributed to less co-localization of LC3 and LAMP2 in GBM cells (Fig. 5b), which was further verified by Co-IP (Fig. 5c). As shown in Fig. 5c, the interaction of LAMP2 with LC3B was decreased after treatment of S670, which indicated the impaired fusion of autophagosome and lysosome. The above findings thus suggested that S670 could block autophagy flux via impairing autophagosome-lysosome fusion, which is similar to CA-5f, an identified late-stage autophagy inhibitor [48].
尽管S670可以启动自噬并诱导溶酶体的生物合成,但p62水平升高表明S670可能会损害自噬降解[ 47 ]。为了检查S670是否影响晚期自噬,在S670处理后检查自噬通量。首先,用S670进一步处理稳定表达mRFP-GFP标记的LC3B的U87和U251细胞,以进一步确认自噬流。结果表明,S670处理后自噬流被阻断,在U87和U251细胞中观察到黄点(自噬体)增加和红点(自溶酶体)减少(图5a ,补充图S6a )。此外,S670处理导致GBM细胞中LC3和LAMP2的共定位减少(图5b ),这通过Co-IP进一步验证(图5c )。如图5c所示,S670处理后LAMP2与LC3B的相互作用减少,这表明自噬体和溶酶体的融合受损。上述研究结果表明,S670可以通过损害自噬体-溶酶体融合来阻断自噬流,这与CA-5f(一种已确定的晚期自噬抑制剂)类似[ 48 ]。
图 5:S670 通过抑制 STX17 损害自噬体-溶酶体融合,从而加剧铁死亡。

a Immunofluorescence analysis was performed to observe the colocalization of GFP-LC3 and mRFP-LC3 puncta in U87 and U251 cells by laser confocal microscopy. b Immunofluorescent double-staining against LC3 (green) and LAMP2 (red) was performed to analyze the co-localization of LC3 and LAMP2. Cell nuclei were stained with DAPI (blue). Representative co-localization signals were analyzed with ImageJ software (1.52 v). c Cells were treated with S670 (6 μM) for 24 h, and whole cell lysate was collected and immunoprecipitation was performed using anti-LAMP2 (in U87 cells) or anti-LC3 antibody (in U251 cells), and the interaction of LC3 and LAMP2 was determined using Co-IP. d Expression of STX17, SNAP29 and VAMP8 was detected by Western blotting after treatment with 0, 4, 8 and 12 μM S670 for 24 h. e The mRNA level of STX17 was detected by RT-qPCR after the treatment of S670 (6 μM). f GBM cells were treated with S670 (6 μM) for 12 h and S670 was washed out or kept until 24 h. The expression of LC3B and STX17 was detected by Western blotting. g ROS level in U87 cells was measured by DCFH-DA after being cotreated with S670 (6 μM) and CQ (10 μM) for 24 h. Scale bar = 200 nm. h The expression of COX2, GPX4 and STX17 in U251 cells was detected by Western blotting after cotreated with S670 (6 μM) and CQ (10 μM) for 24 h. The data were performed in triplicate and shown as the mean ± SD. *P < 0.05 compared to the NC group.
a进行免疫荧光分析,通过激光共聚焦显微镜观察 U87 和 U251 细胞中 GFP-LC3 和 mRFP-LC3 斑点的共定位。 b对 LC3(绿色)和 LAMP2(红色)进行免疫荧光双重染色,以分析 LC3 和 LAMP2 的共定位。细胞核用 DAPI(蓝色)染色。使用 ImageJ 软件 (1.52 v) 分析代表性共定位信号。 c用 S670 (6 μM) 处理细胞 24 小时,收集全细胞裂解液,并使用抗 LAMP2(在 U87 细胞中)或抗 LC3 抗体(在 U251 细胞中)进行免疫沉淀,以及 LC3 和LAMP2 使用 Co-IP 进行测定。 d用0、4、8和12μM S670处理24小时后,通过Western blotting检测STX17、SNAP29和VAMP8的表达。 e S670 (6 μM) 处理后,通过 RT-qPCR 检测 STX17 的 mRNA 水平。 f GBM 细胞用 S670 (6 μM) 处理 12 小时,洗掉 S670 或保留至 24 小时。 Western blotting检测LC3B和STX17的表达。 g U87细胞中ROS水平在与S670(6μM)和CQ(10μM)共处理24小时后通过DCFH-DA测量。比例尺 = 200 nm。 h S670(6 μM)和CQ(10 μM)共处理24 h后,通过Western blotting检测U251细胞中COX2、GPX4和STX17的表达。数据一式三份进行并显示为平均值±SD。 * 与 NC 组相比, P < 0.05。
SNARE complex-mediated fusion is a critical process in autophagosome-lysosome fusion and STX17, SNAP29, and VAMP8 are major factors involved in such a process [49]. Genetic ablation of STX17 and mTOR resulted in over-accumulation of non-fused autophagosomes which elevated ROS generation and conferred cytotoxicity [50]. Thus, we took a further step to investigate whether S670 inhibited the fusion of autophagosome and lysosome by affecting the function of the SNARE complex. It was shown that S670 inhibited the mRNA and protein level of STX17 which was localized on autophagosome in U87 and U251 cells (Fig. 5d, e). However, S670 induced the expression of lysosome-localized VAMP8, which might be explained by the increasing lysosome biogenesis (Fig. 5d), and that S670 had no obvious impact on the expression of SNAP29 (Fig. 5d). To further verify whether inhibition of STX17 leads to autophagosome accumulation, GBM cells were treated with S670 for 12 h and S670 was washed out or kept until 24 h and the expression of LC3B and STX17 were checked. As shown in Fig. 5f, S670 remarkably inhibited the expression of STX17 after treatment for 12 h, which was ahead of the accumulation of LC3B in 24 h (non-washed). Even being washed out with fresh culture at 12 h, the accumulation of LC3B was still increased until 24 h. To further determine whether S670 also affected the assembly ability of SNARE complexes to block autophagosome-lysosome fusion, the interaction of STX17 and VAMP8 was explored by Co-IP (Supplementary Fig. S6b). After the treatment of S670, even the expression of STX17 was decreased, there was no obvious change in the interaction between STX17 and VAMP8, which demonstrated that S670 had no effect on the assembly ability of SNARE complexes. The above results demonstrated that the inhibition of STX17 might contribute to the blockade of autophagosome-lysosome fusion. Collectively, all these results demonstrated that S670 impaired autophagosome-lysosome fusion to block autophagy, which indicated that S670 might be a novel late-stage autophagy inhibitor.
SNARE复合物介导的融合是自噬体-溶酶体融合的关键过程,STX17、SNAP29和VAMP8是参与这一过程的主要因素[ 49 ]。 STX17 和 mTOR 的基因消除导致非融合自噬体过度积累,从而增加 ROS 生成并赋予细胞毒性 [ 50 ]。因此,我们进一步研究S670是否通过影响SNARE复合体的功能来抑制自噬体和溶酶体的融合。结果显示,S670抑制位于U87和U251细胞中自噬体上的STX17的mRNA和蛋白水平(图5d,e )。然而,S670诱导了溶酶体定位的VAMP8的表达,这可能是由于溶酶体生物发生的增加来解释的(图5d ),并且S670对SNAP29的表达没有明显影响(图5d )。为了进一步验证STX17的抑制是否导致自噬体积累,用S670处理GBM细胞12小时,洗掉S670或保留至24小时,检查LC3B和STX17的表达。如图5f所示,S670处理12小时后显着抑制STX17的表达,领先于24小时LC3B的积累(未洗涤)。即使在12小时时用新鲜培养物洗掉,LC3B的积累仍然增加直至24小时。为了进一步确定S670是否也影响SNARE复合物的组装能力以阻止自噬体-溶酶体融合,通过Co-IP探索了STX17和VAMP8的相互作用(补充图S6b )。 S670处理后,即使STX17的表达降低,STX17与VAMP8之间的相互作用也没有明显变化,这表明S670对SNARE复合物的组装能力没有影响。上述结果表明,抑制STX17可能有助于阻断自噬体-溶酶体融合。总的来说,所有这些结果都表明S670破坏自噬体-溶酶体融合从而阻断自噬,这表明S670可能是一种新型的晚期自噬抑制剂。
Inhibition of autophagosome-lysosome fusion by S670 enhanced ROS accumulation which exacerbated ferroptosis in glioma cells
S670 抑制自噬体-溶酶体融合增强 ROS 积累,加剧胶质瘤细胞铁死亡
Modulating lysosomal activity is another effective way to disrupt autophagic flux using a lysosomotropic agent such as chloroquine (CQ) [51]. We had demonstrated that S670 had no effect on lysosomal function. To explore whether simultaneously disturbing lysosomal function could enhance the anti-GBM activity of S670, CQ was combined with S670 to further inhibit autophagic degradation. The combination of S670 with CQ could exacerbate ROS and lipid ROS generation, suggesting that CQ might enhance ferroptosis induced by S670 (Fig. 5g, Supplementary Fig. S6c). And cell viability assay proved that CQ could increase the cytotoxicity of S670 (Supplementary Fig. S6d). Moreover, the combination of CQ and S670 could further promote the expression of COX2 but exacerbated the inhibition of the expression of GPX4 as well as STX17, which confirmed more dramatical ferroptosis occurred when autophagic degradation was further inhibited (Fig. 5h). All in all, these results suggested that further inhibiting the fusion of autophagosome and lysosome by disturbing lysosomal function could synergize with S670 to aggravate ferroptosis, which also proved from the side that the blockade of self-protective autophagy might dampen the elimination of ROS and participate in the occurrence of ferroptosis in GBM cells.
调节溶酶体活性是使用氯喹(CQ)等溶酶体药物破坏自噬流的另一种有效方法[ 51 ]。我们已经证明 S670 对溶酶体功能没有影响。为了探讨同时干扰溶酶体功能是否可以增强S670的抗GBM活性,将CQ与S670联合以进一步抑制自噬降解。 S670与CQ的组合可以加剧ROS和脂质ROS的产生,表明CQ可能增强S670诱导的铁死亡(图5g ,补充图S6c )。细胞活力测定证明CQ可以增加S670的细胞毒性(补充图S6d )。此外,CQ和S670的组合可以进一步促进COX2的表达,但加剧了对GPX4和STX17表达的抑制,这证实当自噬降解进一步受到抑制时,会发生更严重的铁死亡(图5h )。总而言之,这些结果提示,通过扰乱溶酶体功能进一步抑制自噬体与溶酶体的融合,可以与S670协同加重铁死亡,这也从侧面证明了自我保护性自噬的阻断可能会抑制ROS的清除,参与ROS的清除。 GBM 细胞铁死亡的发生。
S670 suppressed tumor growth in a U87 mouse xenograft model
S670 抑制 U87 小鼠异种移植模型中的肿瘤生长
To evaluate the anti-GBM effect of S670 in vivo, a U87 mouse xenograft model was constructed and mice were treated with TMZ (10 mg/kg, i.g. daily), S670 (25 mg/kg, i.g. daily), S670 (50 mg/kg, i.g. daily) or DMSO until the tumor volume reached 1000 mm3 (Fig. 6a). It was shown that there was no obvious body change in each group during administration (Fig. 6b), and that S670 significantly inhibited tumor growth in vivo (Fig. 6c). Moreover, tumor weights in S670 administration groups were significantly decreased (Fig. 6d, e). Immunohistochemistry (IHC) showed that the expression of Ki67 was reduced and the expression of LC3 and LAMP2 was increased in tumor tissues (Fig. 6f, g). Organ index was calculated to confirm the safety of S670 administration and there was no significant difference in those five major organ indexes in each group (Supplementary Fig. S7a–e). The integrity of the main solid organs was assessed by HE staining, and there was no obvious induction of detectable toxicity in the group of high dosage (S670, 50 mg/kg) (Fig. 6h). All in all, these results showed that S670 exhibited prominent anti-tumor activity in vivo with good safety.
为了评估S670的体内抗GBM作用,构建了U87小鼠异种移植模型,并用TMZ(10 mg/kg,每天ig)、S670(25 mg/kg,每天ig)、S670(50 mg)治疗小鼠。 /kg,每天ig)或DMSO直至肿瘤体积达到1000mm 3 (图6a )。结果显示,给药过程中各组均无明显的机体变化(图6b ),并且S670在体内显着抑制肿瘤生长(图6c )。此外,S670给药组的肿瘤重量显着降低(图6d,e )。免疫组织化学(IHC)显示肿瘤组织中Ki67的表达减少,LC3和LAMP2的表达增加(图6f,g )。计算器官指数以确认S670给药的安全性,每组中这五个主要器官指数没有显着差异(补充图S7a-e )。通过HE染色评估主要实体器官的完整性,在高剂量(S670,50mg/kg)组中没有明显诱导可检测到的毒性(图6h )。总而言之,这些结果表明S670在体内表现出显着的抗肿瘤活性,且安全性良好。
图 6:S670 在 U87 小鼠异种移植模型中抑制肿瘤生长。

a Schematic outline of the construction of U87 xenograft tumor model in nude mice and the mice were treated with TMZ (10 mg/kg daily i.g. injection), S670 (25 mg/kg daily i.g. injection), S670 (50 mg/kg daily i.g. injection) (n = 6), or DMSO for the vehicle group. b Changes in body weight in each group during the S670 administration period (n = 6). c Changes in tumor volume in each group during the administration period (n = 6). d Tumor weight was recorded and calculated in each group after the administration period (n = 6). e Image of tumors of the vehicle and different administration groups. f Representative image of the tumors that were fixed and stained with Ki67 in each group with corresponding quantification results. Scale bar = 100 nm. g Representative image of the tumors that were fixed and stained with LC3 and LAMP2 in vehicle group and S670 (50 mg/kg) group. Scale bar = 100 nm. h Representative image of the hearts, livers, spleens, lungs and kidneys from the vehicle group and S670 (50 mg/kg) group that were fixed and stained with H&E. The data were shown as the mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 compared to the control group.
a裸鼠U87异种移植瘤模型构建示意图,小鼠分别给予TMZ(每日10mg/kg ig注射)、S670(每日25mg/kg ig注射)、S670(每日50mg/kg ig注射)注射)( n = 6),或媒介物组为 DMSO。 b S670给药期间各组体重变化( n = 6)。 c给药期间各组肿瘤体积的变化( n = 6)。 d给药期后记录并计算各组的肿瘤重量( n = 6)。 e载体和不同给药组的肿瘤图像。 f每组中固定并用 Ki67 染色的肿瘤的代表性图像以及相应的定量结果。比例尺 = 100 nm。 g在赋形剂组和S670(50mg/kg)组中固定并用LC3和LAMP2染色的肿瘤的代表性图像。比例尺 = 100 nm。 h来自赋形剂组和 S670 (50 mg/kg) 组的心脏、肝脏、脾脏、肺和肾的代表性图像,经 H&E 固定和染色。数据显示为平均值±SD。与对照组相比,* P < 0.05,** P < 0.01,*** P < 0.001,**** P < 0.0001。
Discussion 讨论
GBM is the most common and malignant primary brain tumor and TMZ is the only approved first-line alkylating agent as the gold standard therapy for GBM but a majority of patients have no response to TMZ during treatment [52]. It is important to develop new drugs for GBM. Based on the potent anti-GBM activity of AKBA, a series of novel amide derivatives of AKBA were designed and synthesized and we found the derivative S670 exerted superior anti-GBM activity compared with AKBA and other derivatives. Furthermore, we elaborated on the molecular mechanism of S670 and discovered S670 could induce ferroptosis in GBM cells. In addition, S670 could activate Nrf2-mediated autophagosome biogenesis and TFEB-mediated lysosome biogenesis but block STX17-mediated fusion of autophagosome and lysosome. In vivo, S670 significantly suppressed tumor growth with no obvious adverse effect. Collectively, our results demonstrated S670, an amide derivative of AKBA, is a potential drug candidate for GBM.
GBM 是最常见和恶性的原发性脑肿瘤,TMZ 是唯一被批准作为 GBM 金标准治疗的一线烷化剂,但大多数患者在治疗期间对 TMZ 没有反应[ 52 ]。开发治疗 GBM 的新药非常重要。基于AKBA强大的抗GBM活性,我们设计并合成了一系列新型AKBA酰胺衍生物,我们发现衍生物S670与AKBA和其他衍生物相比具有更优异的抗GBM活性。此外,我们详细阐述了S670的分子机制,发现S670可以诱导GBM细胞铁死亡。此外,S670可以激活Nrf2介导的自噬体生物发生和TFEB介导的溶酶体生物发生,但阻断STX17介导的自噬体和溶酶体的融合。在体内,S670显着抑制肿瘤生长,且无明显副作用。总的来说,我们的结果表明 S670(AKBA 的酰胺衍生物)是 GBM 的潜在候选药物。
Ferroptosis is a new form of regulated cell death driven by an overload of lipid peroxide and mitochondrial ROS is critical for lipid peroxide [5]. Here, we first uncovered ferroptosis as the novel anti-GBM mechanism of S670, which was distinct from AKBA-induced apoptosis in glioma [23]. S670 could induce ferroptosis in GBM cells via stimulating mitochondrial ROS generation, inhibition of GPX4 and activation of COX2. To mitigate such oxidative damage, protective autophagy was activated in glioma cells. S670 could induce ROS generation which activated autophagy by activating Nrf2 and contributed to autophagy initiation. Such autophagy induction was similar to other anti-GBM therapies as a hallmark effect under stress-mediated damage [53]. For example, flavokawain B, a natural kava chalcone, induced protective autophagy by endoplasmic reticulum stress in GBM [54].
铁死亡是一种由脂质过氧化物超载驱动的新型受调节细胞死亡,线粒体 ROS 对于脂质过氧化物至关重要[ 5 ]。在这里,我们首次发现铁死亡是 S670 的新型抗 GBM 机制,这与 AKBA 诱导的胶质瘤细胞凋亡不同[ 23 ]。 S670 可通过刺激线粒体 ROS 生成、抑制 GPX4 和激活 COX2 来诱导 GBM 细胞铁死亡。为了减轻这种氧化损伤,神经胶质瘤细胞中的保护性自噬被激活。 S670可以诱导ROS产生,通过激活Nrf2来激活自噬,从而促进自噬的启动。这种自噬诱导与其他抗 GBM 疗法相似,是应激介导损伤下的标志性效应 [ 53 ]。例如,flavokawain B(一种天然卡瓦查尔酮)通过 GBM 中的内质网应激诱导保护性自噬 [ 54 ]。
Apart from the new discovery of new anti-tumor mechanism of S670, we observed an interesting phenomenon that S670 induced increasing vacuoles in GBM cells. The increasing vacuoles was further demonstrated as increasing and enlarged lysosomes. Disruption of lysosome integrity by inducing lysosomal membrane permeabilization (LMP) could lead to the release of lysosomal contents into cytosol such as cathepsins, which contributed to lysosome-dependent cell death (LDCD). And the enlarged lysosomes were considered the prerequisite of LMP [55]. LMP has been put forward as an effective strategy for cancer therapy, especially leukemia. Hu et al. found that LW-218, a newly synthesized derivative of wogonin, could induce cholesterol-associated lysosomal damage in hematological malignancy cells [42]. We wonder whether such vacuoles originated from increasingly enlarged lysosomes indicated that S670 induced LMP in GBM cells. However, S670 had no impact on the expression of CSTD and lysosomal pH, revealing that S670 did not impair lysosomal proteolytic activity and further excluded the possibility of LMP occurrence after S670 treatment. Thus, we speculated TFEB mediated lysosomal biogenesis might be a cytoprotective mechanism in response to autophagy activation which was validated by TFEB knocking down.
除了S670抗肿瘤新机制的新发现外,我们还观察到一个有趣的现象,即S670诱导GBM细胞中空泡增加。液泡的增加进一步证明了溶酶体的增加和扩大。通过诱导溶酶体膜透化(LMP)破坏溶酶体完整性可能导致溶酶体内容物释放到细胞质中,例如组织蛋白酶,从而导致溶酶体依赖性细胞死亡(LDCD)。溶酶体增大被认为是末次月经发生的先决条件[ 55 ]。 LMP已被提出作为癌症治疗,特别是白血病的有效策略。胡等人。发现LW-218是一种新合成的汉黄芩素衍生物,可诱导血液恶性肿瘤细胞中胆固醇相关的溶酶体损伤[ 42 ]。我们想知道这些液泡是否源自日益增大的溶酶体,表明 S670 在 GBM 细胞中诱导 LMP。然而,S670对CSTD的表达和溶酶体pH没有影响,表明S670没有损害溶酶体蛋白水解活性,进一步排除了S670处理后发生LMP的可能性。因此,我们推测 TFEB 介导的溶酶体生物发生可能是响应自噬激活的细胞保护机制,这一点通过 TFEB 敲低得到验证。
Although S670 induced the biogenesis of autophagosome and lysosome, we surprisingly found the STX17-mediated fusion of autophagosome and lysosome was inhibited by S670. Over-accumulation of non-fused autophagosomes could induce ROS generation which conferred cytotoxicity, which could be achieved by simultaneous ablation of mTOR and STX17 [50]. Such phenomena of autophagosome accumulation had also been illustrated in the mechanism of the anti-cholangiocarcinoma effect of prodigiosin and methamphetamine-induced autophagy-associated neuronal death [56]. Since S670 induced lysosome biogenesis without altering lysosomal function, we wondered whether interfering with lysosomal function could further enhance autophagy blockade caused by S670 to enhance anti-tumor activity. As expected, CQ could exacerbate S670-induced ROS generation and ferroptosis in GBM cells, which provided new insights into chloroquine combination treatment in glioma [57].
尽管S670诱导自噬体和溶酶体的生物发生,但我们令人惊讶地发现STX17介导的自噬体和溶酶体的融合被S670抑制。非融合自噬体的过度积累可能会诱导ROS产生,从而产生细胞毒性,这可以通过同时消融mTOR和STX17来实现[ 50 ]。这种自噬体积累的现象也在灵菌红和甲基苯丙胺诱导的自噬相关神经元死亡的抗胆管癌作用机制中得到了说明[ 56 ]。由于S670诱导溶酶体生物合成而不改变溶酶体功能,我们想知道干扰溶酶体功能是否可以进一步增强S670引起的自噬阻断,从而增强抗肿瘤活性。正如预期的那样,CQ 可以加剧 GBM 细胞中 S670 诱导的 ROS 生成和铁死亡,这为胶质瘤的氯喹联合治疗提供了新的见解[ 57 ]。
TMZ could induce cytoprotecting autophagy by ATM/AMPK/ULK1 pathway, which is one of the critical mechanisms of TMZ resistance in glioma [52]. Although S670 exerted anti-GBM effect in vivo with a higher dosage than TMZ, the special autophagy-blockade effect could avoid cytoprotective autophagy-induced drug resistance in GBM. Moreover, the autophagy modulatory activity of S670 also indicated its potential to overcome temozolomide resistance by inhibiting protective autophagy induced by TMZ [58].
TMZ可以通过ATM/AMPK/ULK1通路诱导细胞保护性自噬,这是胶质瘤中TMZ耐药的关键机制之一[ 52 ]。尽管S670在体内发挥抗GBM作用的剂量高于TMZ,但其特殊的自噬阻断作用可以避免细胞保护性自噬诱导的GBM耐药。此外,S670的自噬调节活性也表明其通过抑制TMZ诱导的保护性自噬来克服替莫唑胺耐药性的潜力[ 58 ]。
In conclusion, compound S670, a novel amide derivative of AKBA, showed potent anti-GBM activity in vitro. S670 could induce mitochondrial ROS generation which contributed to ferroptosis and activation of autophagosome and lysosome biogenesis. Moreover, S670 impaired autophagosome-lysosome fusion to block autophagy flux by inhibiting the expression of SXT17, which exacerbated ROS accumulation in GBM cells to further strengthen the anti-tumor effect of S670 by enhancing ferroptosis in glioma cells. The mechanism of the anti-GBM effect of S670 was summarized in Fig. 7. In addition, S670 exerted a prominent anti-tumor effect in vivo with good safety. Thus, S670 might be a promising drug candidate for GBM.
总之,化合物S670是AKBA的一种新型酰胺衍生物,在体外表现出有效的抗GBM活性。 S670 可以诱导线粒体 ROS 生成,从而导致铁死亡以及自噬体和溶酶体生物发生的激活。此外,S670通过抑制SXT17的表达来破坏自噬体-溶酶体融合,从而阻断自噬流,从而加剧GBM细胞中ROS的积累,从而通过增强胶质瘤细胞中的铁死亡来进一步增强S670的抗肿瘤作用。 S670的抗GBM作用的机制总结于图7中。此外,S670在体内发挥了显着的抗肿瘤作用,且安全性良好。因此,S670 可能是治疗 GBM 的有前途的候选药物。
图 7:S670 抗 GBM 活性的拟议机制方案。

S670 stimulates ROS generation to induce ferroptosis in GBM cells. Moreover, S670 induces autophagosome biogenesis via ROS-activated Nrf2 nuclear translocation and stimulates lysosome biogenesis by ROS-activated TFEB nuclear translocation. However, S670 interrupts the fusion of autophagosome and lysosome to exacerbate ferroptosis by inhibiting the expression of STX17 in GBM cells. Thus, S670 induces ferroptosis in human glioblastoma cells by generating ROS and inhibiting STX17-mediated fusion of autophagosome and lysosome.
S670 刺激 ROS 生成,诱导 GBM 细胞铁死亡。此外,S670 通过 ROS 激活的 Nrf2 核转位诱导自噬体生物发生,并通过 ROS 激活的 TFEB 核转位刺激溶酶体生物发生。然而,S670通过抑制GBM细胞中STX17的表达来中断自噬体和溶酶体的融合,从而加剧铁死亡。因此,S670 通过产生 ROS 并抑制 STX17 介导的自噬体和溶酶体融合来诱导人胶质母细胞瘤细胞铁死亡。
References 参考
Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS Statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro Oncol. 2020;22:iv1–iv96.
奥斯特罗姆 QT、帕蒂尔 N、乔菲 G、韦特 K、克鲁奇科 C、巴恩霍尔茨-斯隆 JS。 CBTRUS统计报告:2013-2017年美国诊断出的原发性脑部和其他中枢神经系统肿瘤。神经肿瘤。 2020;22:iv1–iv96。Weller M, Wick W, Aldape K, Brada M, Berger M, Pfister SM, et al. Glioma. Nat Rev Dis Prim. 2015;1:15017.
Weller M、Wick W、Aldape K、Brada M、Berger M、Pfister SM 等。神经胶质瘤。 Nat Rev Dis Prim。 2015;1:15017。Rong L, Li N, Zhang Z. Emerging therapies for glioblastoma: current state and future directions. J Exp Clin Cancer Res. 2022;41:142.
Rong L,Li N,Zhang Z。胶质母细胞瘤的新兴疗法:现状和未来方向。 J Exp 临床癌症研究杂志。 2022;41:142。van Solinge TS, Nieland L, Chiocca EA, Broekman MLD. Advances in local therapy for glioblastoma - taking the fight to the tumour. Nat Rev Neurol. 2022;18:221–36.
van Solinge TS、Nieland L、Chiocca EA、Broekman MLD。胶质母细胞瘤局部治疗的进展——与肿瘤作斗争。纳特·尼罗尔牧师。 2022;18:221–36。Stockwell BR. Ferroptosis turns 10: emerging mechanisms, physiological functions, and therapeutic applications. Cell. 2022;185:2401–21.
斯托克韦尔 BR。铁死亡十周年:新兴机制、生理功能和治疗应用。细胞。 2022;185:2401–21。Liu T, Zhu C, Chen X, Guan G, Zou C, Shen S, et al. Ferroptosis, as the most enriched programmed cell death process in glioma, induces immunosuppression and immunotherapy resistance. Neuro Oncol. 2022;24:1113–25.
刘涛,朱成,陈X,关刚,邹成,沉S,等。铁死亡是神经胶质瘤中最丰富的程序性细胞死亡过程,可诱导免疫抑制和免疫治疗抵抗。神经肿瘤。 2022;24:1113–25。Shi J, Yang N, Han M, Qiu C. Emerging roles of ferroptosis in glioma. Front Oncol. 2022;12:993316.
Shi J,Yang N,Han M,Qiu C.铁死亡在神经胶质瘤中的新作用。前安科尔。 2022;12:993316。Xia L, Gong M, Zou Y, Wang Z, Wu B, Zhang S, et al. Apatinib induces ferroptosis of glioma cells through modulation of the VEGFR2/Nrf2 pathway. Oxid Med Cell Longev. 2022;2022:9925919.
夏丽,龚明,邹Y,王Z,吴B,张S,等。阿帕替尼通过调节 VEGFR2/Nrf2 通路诱导神经胶质瘤细胞铁死亡。氧化医学细胞长寿。 2022;2022:9925919。Zhang L, Song J, Kong L, Yuan T, Li W, Zhang W, et al. The strategies and techniques of drug discovery from natural products. Pharmacol Ther. 2020;216:107686.
张L,宋J,孔L,袁T,李W,张W,等。从天然产物中发现药物的策略和技术。 Pharmacol Ther。 2020;216:107686。Lu S, Wang XZ, He C, Wang L, Liang SP, Wang CC, et al. ATF3 contributes to brucine-triggered glioma cell ferroptosis via promotion of hydrogen peroxide and iron. Acta Pharmacol Sin. 2021;42:1690–702.
卢 S,王新征,何 C,王 L,梁 SP,王 CC,等。 ATF3 通过促进过氧化氢和铁促进马钱子碱触发的神经胶质瘤细胞铁死亡。药学学报。 2021;42:1690–702。Zhan S, Lu L, Pan SS, Wei XQ, Miao RR, Liu XH, et al. Targeting NQO1/GPX4-mediated ferroptosis by plumbagin suppresses in vitro and in vivo glioma growth. Br J Cancer. 2022;127:364–76.
詹S,卢L,潘SS,魏XQ,苗RR,刘XH,等。白花丹素靶向 NQO1/GPX4 介导的铁死亡可抑制体外和体内神经胶质瘤的生长。 Br J 癌症。 2022;127:364–76。Wang Z, Ding Y, Wang X, Lu S, Wang C, He C, et al. Pseudolaric acid B triggers ferroptosis in glioma cells via activation of Nox4 and inhibition of xCT. Cancer Lett. 2018;428:21–33.
王Z,丁Y,王X,卢S,王C,何C,等。 Pseudolaric Acid B 通过激活 Nox4 和抑制 xCT 来触发神经胶质瘤细胞中的铁死亡。癌症快报。 2018;428:21-33。Chen TC, Chuang JY, Ko CY, Kao TJ, Yang PY, Yu CH, et al. AR ubiquitination induced by the curcumin analog suppresses growth of temozolomide-resistant glioblastoma through disrupting GPX4-mediated redox homeostasis. Redox Biol. 2020;30:101413.
陈TC,庄JY,柯CY,高TJ,杨PY,于CH,等。姜黄素类似物诱导的 AR 泛素化通过破坏 GPX4 介导的氧化还原稳态来抑制替莫唑胺耐药的胶质母细胞瘤的生长。氧化还原生物。 2020;30:101413。Feng Y, He D, Yao Z, Klionsky DJ. The machinery of macroautophagy. Cell Res. 2014;24:24–41.
冯Y,何东,姚Z,Klionsky DJ。巨自噬机制。细胞研究。 2014;24:24–41。Onorati AV, Dyczynski M, Ojha R, Amaravadi RK. Targeting autophagy in cancer. Cancer. 2018;124:3307–18.
Onorati AV、Dyczynski M、Ojha R、Amaravadi RK。靶向癌症中的自噬。癌症。 2018;124:3307–18。Ishaq M, Ojha R, Sharma AP, Singh SK. Autophagy in cancer: recent advances and future directions. Semin Cancer Biol. 2020;66:171–81.
伊沙克·M、奥贾·R、夏尔马·AP、辛格·SK。癌症中的自噬:最新进展和未来方向。塞明癌症生物学。 2020;66:171–81。Taylor MA, Das BC, Ray SK. Targeting autophagy for combating chemoresistance and radioresistance in glioblastoma. Apoptosis. 2018;23:563–75.
泰勒 MA、达斯 BC、雷 SK。靶向自噬以对抗胶质母细胞瘤的化学耐药性和放射耐药性。细胞凋亡。 2018;23:563–75。Sanati M, Binabaj MM, Ahmadi SS, Aminyavari S, Javid H, Mollazadeh H, et al. Recent advances in glioblastoma multiforme therapy: a focus on autophagy regulation. Biomed Pharmacother. 2022;155:113740.
Sanati M、Binabaj MM、Ahmadi SS、Aminyavari S、Javid H、Mollazadeh H 等。多形性胶质母细胞瘤治疗的最新进展:关注自噬调节。生物医学药剂师。 2022;155:113740。Siddiqui MZ. Boswellia serrata, a potential antiinflammatory agent: an overview. Indian J Pharm Sci. 2011;73:255–61.
西迪基 MZ.齿叶乳香,一种潜在的抗炎剂:概述。印度医药科学杂志。 2011;73:255–61。Efferth T, Oesch F. Anti-inflammatory and anti-cancer activities of frankincense: targets, treatments and toxicities. Semin Cancer Biol. 2022;80:39–57.
Efferth T,Oesch F。乳香的抗炎和抗癌活性:目标、治疗和毒性。塞明癌症生物学。 2022;80:39–57。Siddiqui A, Shah Z, Jahan RN, Othman I, Kumari Y. Mechanistic role of boswellic acids in Alzheimer’s disease: emphasis on anti-inflammatory properties. Biomed Pharmacother. 2021;144:112250.
Siddiqui A、Shah Z、Jahan RN、Othman I、Kumari Y。乳香酸在阿尔茨海默病中的机制作用:强调抗炎特性。生物医学药剂师。 2021;144:112250。Gong Y, Jiang X, Yang S, Huang Y, Hong J, Ma Y, et al. The biological activity of 3-O-acetyl-11-keto-β-boswellic acid in nervous system diseases. Neuromolecular Med. 2022;24:374–84.
龚Y,蒋X,杨S,黄Y,洪J,马Y,等。 3-O-乙酰基-11-酮-β-乳香酸在神经系统疾病中的生物活性。神经分子医学。 2022;24:374–84。Li W, Liu J, Fu W, Zheng X, Ren L, Liu S, et al. 3-O-acetyl-11-keto-β-boswellic acid exerts anti-tumor effects in glioblastoma by arresting cell cycle at G2/M phase. J Exp Clin Cancer Res. 2018;37:132.
Li W, Ren L, Zheng X, Liu J, Wang J, Ji T, et al. 3-O-Acetyl-11-keto- β -boswellic acid ameliorated aberrant metabolic landscape and inhibited autophagy in glioblastoma. Acta Pharm Sin B. 2020;10:301–12.
Mendonsa AM, Na TY, Gumbiner BM. E-cadherin in contact inhibition and cancer. Oncogene. 2018;37:4769–80.
Cao ZQ, Wang Z, Leng P. Aberrant N-cadherin expression in cancer. Biomed Pharmacother. 2019;118:109320.
Marefati N, Beheshti F, Memarpour S, Bayat R, Naser Shafei M, Sadeghnia HR, et al. The effects of acetyl-11-keto-β-boswellic acid on brain cytokines and memory impairment induced by lipopolysaccharide in rats. Cytokine. 2020;131:155107.
Yang Y, Karakhanova S, Hartwig W, D’Haese JG, Philippov PP, Werner J, et al. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. 2016;231:2570–81.
Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843.
Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol. 2021;220:e202105043.
Zhao C, Yu D, He Z, Bao L, Feng L, Chen L, et al. Endoplasmic reticulum stress-mediated autophagy activation is involved in cadmium-induced ferroptosis of renal tubular epithelial cells. Free Radic Biol Med. 2021;175:236–48.
Chen X, Yu C, Kang R, Kroemer G, Tang D. Cellular degradation systems in ferroptosis. Cell Death Differ. 2021;28:1135–48.
Zhou J, Li XY, Liu YJ, Feng J, Wu Y, Shen HM, et al. Full-coverage regulations of autophagy by ROS: from induction to maturation. Autophagy. 2022;18:1240–55.
Katsuragi Y, Ichimura Y, Komatsu M. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 2015;282:4672–8.
Hill SM, Wrobel L, Rubinsztein DC. Post-translational modifications of Beclin 1 provide multiple strategies for autophagy regulation. Cell Death Differ. 2019;26:617–29.
Xu F, Hua C, Tautenhahn HM, Dirsch O, Dahmen U. The role of autophagy for the regeneration of the aging liver. Int J Mol Sci. 2020;21:3606.
Lu Z, Ren Y, Yang L, Jia A, Hu Y, Zhao Y, et al. Inhibiting autophagy enhances sulforaphane-induced apoptosis via targeting NRF2 in esophageal squamous cell carcinoma. Acta Pharm Sin B. 2021;11:1246–60.
Sharma G, Guardia CM, Roy A, Vassilev A, Saric A, Griner LN, et al. A family of PIKFYVE inhibitors with therapeutic potential against autophagy-dependent cancer cells disrupt multiple events in lysosome homeostasis. Autophagy. 2019;15:1694–718.
Saftig P, Haas A. Turn up the lysosome. Nat Cell Biol. 2016;18:1025–7.
Eskelinen EL. Roles of LAMP-1 and LAMP-2 in lysosome biogenesis and autophagy. Mol Asp Med. 2006;27:495–502.
Zhao C, Qiu S, He J, Peng Y, Xu H, Feng Z, et al. Prodigiosin impairs autophagosome-lysosome fusion that sensitizes colorectal cancer cells to 5-fluorouracil-induced cell death. Cancer Lett. 2020;481:15–23.
Hu P, Li H, Sun W, Wang H, Yu X, Qing Y, et al. Cholesterol-associated lysosomal disorder triggers cell death of hematological malignancy: Dynamic analysis on cytotoxic effects of LW-218. Acta Pharm Sin B. 2021;11:3178–92.
Peng P, Jia D, Cao L, Lu W, Liu X, Liang C, et al. Akebia saponin E, as a novel PIKfyve inhibitor, induces lysosome-associated cytoplasmic vacuolation to inhibit proliferation of hepatocellular carcinoma cells. J Ethnopharmacol. 2021;266:113446.
Hossain MI, Marcus JM, Lee JH, Garcia PL, Singh V, Shacka JJ, et al. Restoration of CTSD (cathepsin D) and lysosomal function in stroke is neuroprotective. Autophagy. 2021;17:1330–48.
Hinton T, Johnston GAR. GABA-enriched teas as neuro-nutraceuticals. Neurochem Int. 2020;141:104895.
Zhang X, Cheng X, Yu L, Yang J, Calvo R, Patnaik S, et al. MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat Commun. 2016;7:12109.
Fu R, Deng Q, Zhang H, Hu X, Li Y, Liu Y, et al. A novel autophagy inhibitor berbamine blocks SNARE-mediated autophagosome-lysosome fusion through upregulation of BNIP3. Cell Death Dis. 2018;9:243.
Zhang L, Qiang P, Yu J, Miao Y, Chen Z, Qu J, et al. Identification of compound CA-5f as a novel late-stage autophagy inhibitor with potent anti-tumor effect against non-small cell lung cancer. Autophagy. 2019;15:391–406.
Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy. 2021;17:2680–8.
Button RW, Roberts SL, Willis TL, Hanemann CO, Luo S. Accumulation of autophagosomes confers cytotoxicity. J Biol Chem. 2017;292:13599–614.
Whitmarsh-Everiss T, Laraia L. Small molecule probes for targeting autophagy. Nat Chem Biol. 2021;17:653–64.
Tomar MS, Kumar A, Srivastava C, Shrivastava A. Elucidating the mechanisms of temozolomide resistance in gliomas and the strategies to overcome the resistance. Biochim Biophys Acta Rev Cancer. 2021;1876:188616.
Ulasov I, Fares J, Timashev P, Lesniak MS. Editing cytoprotective autophagy in glioma: an unfulfilled potential for therapy. Trends Mol Med. 2020;26:252–62.
Wang J, Qi Q, Zhou W, Feng Z, Huang B, Chen A, et al. Inhibition of glioma growth by flavokawain B is mediated through endoplasmic reticulum stress induced autophagy. Autophagy. 2018;14:2007–22.
Wang F, Gómez-Sintes R, Boya P. Lysosomal membrane permeabilization and cell death. Traffic. 2018;19:918–31.
Xu H, Zhu Y, Chen X, Yang T, Wang X, Song X, et al. Mystery of methamphetamine-induced autophagosome accumulation in hippocampal neurons: loss of syntaxin 17 in defects of dynein-dynactin driving and autophagosome-late endosome/lysosome fusion. Arch Toxicol. 2021;95:3263–84.
Compter I, Eekers DBP, Hoeben A, Rouschop KMA, Reymen B, Ackermans L, et al. Chloroquine combined with concurrent radiotherapy and temozolomide for newly diagnosed glioblastoma: a phase IB trial. Autophagy. 2021;17:2604–12.
Jiapaer S, Furuta T, Tanaka S, Kitabayashi T, Nakada M. Potential strategies overcoming the temozolomide resistance for glioblastoma. Neurol Med Chir. 2018;58:405–21.
Acknowledgements
This work was supported by Beijing Natural Science Foundation (7212157). This work was also supported by CAMS Innovation Fund for Medical Sciences (2021-I2M-1-029 and 2022-I2M-JB-011), National Natural Science Foundation of China (81703536, 82073311), Natural Science Foundation of Sichuan Province (2022JDTD0025). We thank Biorender (https://app.biorender.com/) and Figdraw (https://www.figdraw.com/) for the assistance of figure drawing.
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Yang, Yh., Li, W., Ren, Lw. et al. S670, an amide derivative of 3-O-acetyl-11-keto-β-boswellic acid, induces ferroptosis in human glioblastoma cells by generating ROS and inhibiting STX17-mediated fusion of autophagosome and lysosome. Acta Pharmacol Sin 45, 209–222 (2024). https://doi.org/10.1038/s41401-023-01157-9IF: 6.9 Q1
Received:
Accepted:
Published:
Issue Date: