
Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability
ADC毒性的机制和提高ADC耐受性的策略
by

作者:Toan D. Nguyen 、Brandon M. Bordeau 和 Joseph P. Balthasar *
Toan D. Nguyen
,
Brandon M. Bordeau
and
Joseph P. Balthasar
*作者:Toan D. Nguyen 、Brandon M. Bordeau 和 Joseph P. Balthasar *
Department of Pharmaceutical Sciences, University at Buffalo, Buffalo, NY 14214, USA
布法罗大学药学系, 布法罗, 纽约州 14214, 美国
布法罗大学药学系, 布法罗, 纽约州 14214, 美国
*
Author to whom correspondence should be addressed.
应向其发送信件的作者。
应向其发送信件的作者。
Cancers 2023, 15(3), 713; https://doi.org/10.3390/cancers15030713
癌症 2023, 15(3), 713;https://doi.org/10.3390/cancers15030713
癌症 2023, 15(3), 713;https://doi.org/10.3390/cancers15030713
Submission received: 23 December 2022
/
Revised: 19 January 2023
/
Accepted: 19 January 2023
/
Published: 24 January 2023
收到提交日期: 2022年12月23日 / 修回日期: 2023年1月19日 / 录用日期: 2023年1月19日 / 出版日期: 2023年1月24日
收到提交日期: 2022年12月23日 / 修回日期: 2023年1月19日 / 录用日期: 2023年1月19日 / 出版日期: 2023年1月24日
Simple Summary 简单总结
Antibody-drug conjugates (ADC) are a rapidly expanding class of anti-cancer drugs, with twelve agents in current clinical use. Despite recent successes, many ADCs fail during clinical development due to excessive toxicities and unfavorable risk-benefit profiles. Even for those ADCs that have been approved for clinical use, a substantial fraction of treated patients require dose reduction, treatment delays, or treatment discontinuation due to intolerable ADC-associated toxicity. In this report, we review the mechanisms contributing to the clinical toxicity of ADCs, and we discuss strategies to improve ADC tolerability.
抗体-药物偶联物 (ADC) 是一类快速扩展的抗癌药物,目前临床上有 12 种药物。尽管最近取得了成功,但由于过度的毒性和不利的风险收益状况,许多ADC在临床开发过程中失败了。即使对于那些已获准用于临床的ADC,由于无法耐受的ADC相关毒性,很大一部分接受治疗的患者也需要减少剂量、延迟治疗或停止治疗。在本报告中,我们回顾了导致ADC临床毒性的机制,并讨论了提高ADC耐受性的策略。
抗体-药物偶联物 (ADC) 是一类快速扩展的抗癌药物,目前临床上有 12 种药物。尽管最近取得了成功,但由于过度的毒性和不利的风险收益状况,许多ADC在临床开发过程中失败了。即使对于那些已获准用于临床的ADC,由于无法耐受的ADC相关毒性,很大一部分接受治疗的患者也需要减少剂量、延迟治疗或停止治疗。在本报告中,我们回顾了导致ADC临床毒性的机制,并讨论了提高ADC耐受性的策略。
Abstract 抽象
Anti-cancer antibody-drug conjugates (ADCs) aim to expand the therapeutic index of traditional chemotherapy by employing the targeting specificity of monoclonal antibodies (mAbs) to increase the efficiency of the delivery of potent cytotoxic agents to malignant cells. In the past three years, the number of ADCs approved by the Food and Drug Administration (FDA) has tripled. Although several ADCs have demonstrated sufficient efficacy and safety to warrant FDA approval, the clinical use of all ADCs leads to substantial toxicity in treated patients, and many ADCs have failed during clinical development due to their unacceptable toxicity profiles. Analysis of the clinical data has demonstrated that dose-limiting toxicities (DLTs) are often shared by different ADCs that deliver the same cytotoxic payload, independent of the antigen that is targeted and/or the type of cancer that is treated. DLTs are commonly associated with cells and tissues that do not express the targeted antigen (i.e., off-target toxicity), and often limit ADC dosage to levels below those required for optimal anti-cancer effects. In this manuscript, we review the fundamental mechanisms contributing to ADC toxicity, we summarize common ADC treatment-related adverse events, and we discuss several approaches to mitigating ADC toxicity.
抗癌抗体偶联物(ADCs)旨在利用单克隆抗体(mAbs)的靶向特异性来提高强效细胞毒性药物向恶性细胞递送的效率,从而扩大传统化疗的治疗指数。在过去三年中,美国食品和药物管理局(FDA)批准的ADC数量增加了两倍。尽管一些ADC已经显示出足够的疗效和安全性,以保证FDA的批准,但所有ADC的临床使用都会对接受治疗的患者产生实质性的毒性,并且许多ADC由于其不可接受的毒性特征而在临床开发过程中失败。对临床数据的分析表明,剂量限制性毒性 (DLT) 通常由不同的 ADC 共享,这些 ADC 提供相同的细胞毒性有效载荷,与靶向抗原和/或治疗的癌症类型无关。DLT通常与不表达靶向抗原的细胞和组织有关(即脱靶毒性),并且通常将ADC剂量限制在低于最佳抗癌效果所需水平的水平。在这篇手稿中,我们回顾了导致ADC毒性的基本机制,总结了常见的ADC治疗相关不良事件,并讨论了减轻ADC毒性的几种方法。
抗癌抗体偶联物(ADCs)旨在利用单克隆抗体(mAbs)的靶向特异性来提高强效细胞毒性药物向恶性细胞递送的效率,从而扩大传统化疗的治疗指数。在过去三年中,美国食品和药物管理局(FDA)批准的ADC数量增加了两倍。尽管一些ADC已经显示出足够的疗效和安全性,以保证FDA的批准,但所有ADC的临床使用都会对接受治疗的患者产生实质性的毒性,并且许多ADC由于其不可接受的毒性特征而在临床开发过程中失败。对临床数据的分析表明,剂量限制性毒性 (DLT) 通常由不同的 ADC 共享,这些 ADC 提供相同的细胞毒性有效载荷,与靶向抗原和/或治疗的癌症类型无关。DLT通常与不表达靶向抗原的细胞和组织有关(即脱靶毒性),并且通常将ADC剂量限制在低于最佳抗癌效果所需水平的水平。在这篇手稿中,我们回顾了导致ADC毒性的基本机制,总结了常见的ADC治疗相关不良事件,并讨论了减轻ADC毒性的几种方法。
1. Introduction 1. 引言
Antibody-drug conjugates (ADCs) are a rapidly growing class of anti-cancer therapeutics, with more than 100 ADCs undergoing clinical investigation [1]. Currently, 12 ADCs have been approved by the United States Food and Drug Administration (FDA), including gemtuzumab ozogamicin (Mylotarg), brentuximab vedotin (Adcetris), inotuzumab ozogamicin (Besponsa), trastuzumab emtansine (Kadcyla), polatuzumab vedotin (Polivy), enfortumab vedotin (Padcev), trastuzumab deruxtecan (Enhertu), sacituzumab govitecan (Trodelvy), belantamab mafodotin (Blenrep), loncastuximab tesirine (Zynlonta), tisotumab vedotin (Tivdak), and mirvetuximab soravtansine (Elahere).
抗体-药物偶联物(ADC)是一类快速增长的抗癌疗法,目前有100多种ADC正在进行临床研究[1]。目前,美国食品药品监督管理局(FDA)已批准12种ADC,包括吉妥珠单抗奥佐米星(Mylotarg)、布伦妥昔单抗(Adcetris)、伊诺妥珠单抗奥佐米星(Besponsa)、曲妥珠单抗(Kadcyla)、波拉妥珠单抗(Polivy)、恩福妥单抗(Padcev)、曲妥珠单抗deruxtecan(Enhertu)、sacituzumab govitecan(Trodelvy)、belantamab mafodotin(Blenrep)、loncastuximab tesirine(Zynlonta)、tisotumab vedotin(Tivdak)和mirvetuximab soravtansine(Elahere)。
抗体-药物偶联物(ADC)是一类快速增长的抗癌疗法,目前有100多种ADC正在进行临床研究[1]。目前,美国食品药品监督管理局(FDA)已批准12种ADC,包括吉妥珠单抗奥佐米星(Mylotarg)、布伦妥昔单抗(Adcetris)、伊诺妥珠单抗奥佐米星(Besponsa)、曲妥珠单抗(Kadcyla)、波拉妥珠单抗(Polivy)、恩福妥单抗(Padcev)、曲妥珠单抗deruxtecan(Enhertu)、sacituzumab govitecan(Trodelvy)、belantamab mafodotin(Blenrep)、loncastuximab tesirine(Zynlonta)、tisotumab vedotin(Tivdak)和mirvetuximab soravtansine(Elahere)。
ADCs are composed of a monoclonal antibody (mAb) tethered to a cytotoxic small-molecule drug (i.e., “payload”) through a chemical linker. Most of the ADCs that have been investigated have employed payload molecules that have shown poor efficacy and substantial toxicity when administered as unconjugated (i.e., “free”) agents [2,3]. As anticipated by the pharmacokinetics and biodistribution of monoclonal antibody drugs [4,5], where high-affinity mAb binding to cell membrane proteins enables the localization of a substantial fraction of mAb to the targeted cell populations, the chemical conjugation of the payload to the anti-cancer mAb increases the selectivity of the delivery of the payload to the cancer cells, and thereby increases the therapeutic index of the payload [6]. However, despite recent successes, the clinical development of ADCs has been associated with a high failure rate, as off-site toxicity remains problematic, limiting tolerable ADC doses to levels below those required for substantial anti-cancer efficacy [7,8,9,10]. Even for the ADCs that have gained FDA approval, a significant fraction of treated patients require supportive treatment to reduce the severity of ADC-associated toxicities, and many patients require dose reduction, treatment delays, or treatment discontinuation [11].
ADC由单克隆抗体(mAb)组成,该抗体通过化学接头与细胞毒性小分子药物(即“有效载荷”)相连。大多数已研究的ADC都采用了有效载荷分子,这些分子在作为非偶联(即“游离”)试剂给药时显示出较差的疗效和实质性的毒性[2,3]。正如单克隆抗体药物的药代动力学和生物分布所预测的那样 [ 4, 5],其中与细胞膜蛋白结合的高亲和力 mAb 能够将相当一部分 mAb 定位到靶细胞群中,有效载荷与抗癌 mAb 的化学偶联增加了有效载荷递送至癌细胞的选择性, 从而增加有效载荷的治疗指数[6]。然而,尽管最近取得了成功,但ADC的临床开发仍与高失败率有关,因为场外毒性仍然存在问题,将可耐受的ADC剂量限制在低于实质性抗癌疗效所需的水平[7,8,9,10]。即使对于已获得FDA批准的ADC,也有相当一部分接受治疗的患者需要支持治疗以减轻ADC相关毒性的严重程度,许多患者需要减少剂量、延迟治疗或停止治疗[11]。
ADC由单克隆抗体(mAb)组成,该抗体通过化学接头与细胞毒性小分子药物(即“有效载荷”)相连。大多数已研究的ADC都采用了有效载荷分子,这些分子在作为非偶联(即“游离”)试剂给药时显示出较差的疗效和实质性的毒性[2,3]。正如单克隆抗体药物的药代动力学和生物分布所预测的那样 [ 4, 5],其中与细胞膜蛋白结合的高亲和力 mAb 能够将相当一部分 mAb 定位到靶细胞群中,有效载荷与抗癌 mAb 的化学偶联增加了有效载荷递送至癌细胞的选择性, 从而增加有效载荷的治疗指数[6]。然而,尽管最近取得了成功,但ADC的临床开发仍与高失败率有关,因为场外毒性仍然存在问题,将可耐受的ADC剂量限制在低于实质性抗癌疗效所需的水平[7,8,9,10]。即使对于已获得FDA批准的ADC,也有相当一部分接受治疗的患者需要支持治疗以减轻ADC相关毒性的严重程度,许多患者需要减少剂量、延迟治疗或停止治疗[11]。
The investigation of the safety profile of an ADC under development requires a series of preclinical and clinical studies; however, prior ADC development efforts suggest that the clinical toxicity profile of ADCs primarily relates to the payload component [12]. Since a relatively small group of payload molecules (i.e., MMAE, MMAF, DM1, DM4, calicheamicin, SN38, Dxd, PBD) are employed within the vast majority of approved ADCs and ADCs under development [8,13,14,15], consideration of the mechanisms associated with the known toxicities of previously developed ADCs may inform the development of new ADCs. In this manuscript, we provide an overview of the main mechanisms underlying ADC toxicity, and we provide a summary of the clinical safety profile of approved ADCs. Additionally, the manuscript discusses approaches to mitigating or preventing ADC toxicities.
对正在开发的ADC的安全性进行研究需要一系列临床前和临床研究;然而,先前的ADC开发工作表明,ADC的临床毒性特征主要与有效载荷成分有关[12]。由于在绝大多数已获批的ADC和正在开发的ADC中都采用了相对较小的有效载荷分子(即MMAE、MMAF、DM1、DM4、calicheamicin、SN38、Dxd、PBD)[8,13,14,15],因此考虑与先前开发的ADC的已知毒性相关的机制可能会为新ADC的开发提供信息。在这份手稿中,我们概述了ADC毒性的主要机制,并总结了已批准的ADC的临床安全性。此外,该手稿还讨论了减轻或预防ADC毒性的方法。
对正在开发的ADC的安全性进行研究需要一系列临床前和临床研究;然而,先前的ADC开发工作表明,ADC的临床毒性特征主要与有效载荷成分有关[12]。由于在绝大多数已获批的ADC和正在开发的ADC中都采用了相对较小的有效载荷分子(即MMAE、MMAF、DM1、DM4、calicheamicin、SN38、Dxd、PBD)[8,13,14,15],因此考虑与先前开发的ADC的已知毒性相关的机制可能会为新ADC的开发提供信息。在这份手稿中,我们概述了ADC毒性的主要机制,并总结了已批准的ADC的临床安全性。此外,该手稿还讨论了减轻或预防ADC毒性的方法。
2. Mechanisms of ADC Toxicity
2. ADC毒性的机理
It is approximated that only ~0.1% of the injected dose of an ADC is delivered to the targeted diseased cell population, with the vast majority of the administered dose catabolized “off-site” within non-targeted healthy cells, potentially leading to unwanted toxicities [16,17]. Off-site ADC toxicity may be categorized as “on-target” or “off-target”, where on-target toxicity proceeds through ADC binding to the targeted cell surface protein on healthy cells. Each component of the ADC, including the antibody, linker, and payload, may affect the extent of the ADC-induced toxicities. In this section, several mechanisms that lead to the toxicities of ADCs are discussed (Figure 1).
据估计,只有~0.1%的注射剂量的ADC被递送至靶向病变细胞群,绝大多数给药剂量在非靶向健康细胞内“异位”分解代谢,可能导致不必要的毒性[16,17]。场外ADC毒性可分为“靶向”或“脱靶”,其中靶向毒性通过ADC与健康细胞上的靶向细胞表面蛋白结合进行。ADC的每个组分,包括抗体、接头和有效载荷,都可能影响ADC诱导的毒性程度。在本节中,讨论了导致ADC毒性的几种机制(图1)。
据估计,只有~0.1%的注射剂量的ADC被递送至靶向病变细胞群,绝大多数给药剂量在非靶向健康细胞内“异位”分解代谢,可能导致不必要的毒性[16,17]。场外ADC毒性可分为“靶向”或“脱靶”,其中靶向毒性通过ADC与健康细胞上的靶向细胞表面蛋白结合进行。ADC的每个组分,包括抗体、接头和有效载荷,都可能影响ADC诱导的毒性程度。在本节中,讨论了导致ADC毒性的几种机制(图1)。

Figure 1.
Mechanisms of ADC toxicity. Uptake of intact ADCs into normal cells may occur through non-specific endocytosis, or through internalization upon binding to the target antigen or to Fc/C-type lectin receptors. Payloads released from ADC deconjugation or other targeted/non-targeted apoptotic cells in the extracellular fluid may also enter normal cells via passive diffusion for membrane-permeable payloads or via non-specific endocytosis for membrane-impermeable linker-payload adducts. Created with BioRender.com.
图 1.ADC毒性的机制。完整ADCs通过非特异性内吞作用或通过与靶抗原或Fc/C型凝集素受体结合时的内化而被正常细胞摄取。从细胞外液中ADC解偶联或其他靶向/非靶向凋亡细胞释放的有效载荷也可以通过膜渗透性有效载荷的被动扩散或通过膜不可渗透的连接子有效载荷加合物的非特异性内吞作用进入正常细胞。用 BioRender.com 创建。
图 1.ADC毒性的机制。完整ADCs通过非特异性内吞作用或通过与靶抗原或Fc/C型凝集素受体结合时的内化而被正常细胞摄取。从细胞外液中ADC解偶联或其他靶向/非靶向凋亡细胞释放的有效载荷也可以通过膜渗透性有效载荷的被动扩散或通过膜不可渗透的连接子有效载荷加合物的非特异性内吞作用进入正常细胞。用 BioRender.com 创建。
2.1. Target-Independent Toxicity: Off-Target, Off-Site Toxicity
2.1. 靶标无关毒性:脱靶、异地毒性
Conceptually, ADCs are expected to enhance the selectivity of chemotherapy by facilitating the targeted delivery of cytotoxic payload molecules to the desired cell populations (on-target, on-site toxicity) while decreasing the delivery of the payloads to non-targeted healthy tissues, thus widening the therapeutic index. The anticipated safety concern for anti-cancer ADCs during the early phases of technology development was on-target (i.e., target-mediated) toxicity within tissues with some degree of expression of the target antigen, and the differential expression of the target in cancer cells versus in healthy tissues was expected to be the critical determinant of the therapeutic index of the ADCs [18]. However, clinical experience with ADCs subsequently demonstrated that the dose-limiting toxicities (DLTs) are rarely driven by target expression in healthy tissues. In a detailed review of preclinical and clinical data from 20 investigational new drug (IND) applications for ADCs filed between 2012 and 2013, the authors found that ADCs with the same class of linker/payload typically shared highly similar toxicity profiles, DLTs, and maximum tolerated doses (MTDs), regardless of the antigen targeted and regardless of the extent of the antigen’s expression in healthy tissues [15]. For example, the review discussed eight different ADCs under clinical development that incorporated the same vc-MMAE linker-payload composition. Each MMAE ADC was advanced to Phase II clinical investigation at a very similar dose (i.e., within the narrow range of 1.8 mg/kg to 2.4 mg/kg). All eight MMAE ADCs demonstrated similar DLTs (severe bone marrow toxicity, sepsis, and severe motor neuropathy). This same toxicity profile is shown for all FDA-approved vc-MMAE ADCs, including polatuzumab vedotin [19], enfortumab vedotin [20], and tisotumab vedotin [21]. Similarly, in a review of clinical ADC data published between 2010 and 2014, Masters et al. found that the prevalent grade 3/4 toxicities associated ADCs were consistent with their payload class [12]. For instance, severe anemia, neutropenia, and peripheral neuropathy were commonly reported for MMAE ADCs. For DM1 ADCs, grade 3/4 thrombocytopenia and hepatic toxicity are typically observed, and severe ocular toxicity is consistently reported for MMAF and DM4 ADCs. Recently, Saber and Leighton performed a follow-up analysis of 15 IND applications for ADCs containing PBD-dimer payloads submitted between 2013 and 2017. They found that the toxicity profiles of the PBD-ADCs were highly comparable, with common adverse events including vascular leak syndrome, elevated liver enzymes, bone marrow suppression, gastrointestinal events, metabolic effects, musculoskeletal events, neuropathy, pain, dyspnea, fatigue, and kidney injury [22]. The observations found in these studies were consistent with the findings from a recent systemic review and meta-analysis conducted by Zhu et. al. on common and severe treatment-related adverse events associated with ADCs in clinical trials between 2000 and 2022 [23]. These findings suggest that, to date, most of the off-site ADC toxicity relates to off-target delivery of the cytotoxic payload, and that off-target payload delivery is the critical driver for the tolerability of ADCs and, ultimately, the recommended doses used in patients. Of course, it is not completely unexpected that on-target toxicity is infrequently a major concern during the clinical evaluation and use of ADCs. Agents leading to severe on-target toxicity may be likely to be identified quickly during the early phases of preclinical development, leading to deselection prior to their advancement to clinical studies.
从概念上讲,ADC有望通过促进细胞毒性有效载荷分子靶向递送至所需细胞群(靶向,现场毒性)来增强化疗的选择性,同时减少有效载荷向非靶向健康组织的递送,从而扩大治疗指数。在技术开发的早期阶段,抗癌ADC的预期安全性问题是靶向(即靶向介导)在具有一定程度靶抗原表达的组织中的毒性,靶标在癌细胞与健康组织中的差异表达预计将是ADC治疗指数的关键决定因素[18]。然而,随后ADC的临床经验表明,剂量限制性毒性(DLT)很少由健康组织中的靶表达驱动。在对2012年至2013年间提交的20项ADC研究性新药(IND)申请的临床前和临床数据进行详细审查时,作者发现,无论靶向抗原和抗原在健康组织中的表达程度如何,具有相同类别的接头/有效载荷的ADC通常具有高度相似的毒性特征、DLT和最大耐受剂量(MTD)[15]。例如,该综述讨论了八种正在临床开发的不同ADC,它们包含相同的vc-MMAE连接子-有效载荷组成。每个MMAE ADC都以非常相似的剂量(即在1.8 mg/kg至2.4 mg/kg的狭窄范围内)进入II期临床研究。所有 8 种 MMAE ADC 都表现出相似的 DLT(严重骨髓毒性、脓毒症和严重运动神经病变)。 所有FDA批准的vc-MMAE ADC都显示出相同的毒性特征,包括polatuzumab vedotin [ 19]、enfortumab vedotin [ 20]和tisotumab vedotin [ 21]。同样,在对2010-2014年间发表的临床ADC数据的回顾中,Masters等人发现,与ADC相关的普遍3/4级毒性与其有效载荷等级一致[12]。例如,MMAE ADC 通常报告严重贫血、中性粒细胞减少和周围神经病变。对于 DM1 ADC,通常观察到 3/4 级血小板减少症和肝毒性,而 MMAF 和 DM4 ADC 的严重眼毒性报告一致。最近,Saber和Leighton对2013年至2017年间提交的15份含有PBD-二聚体有效载荷的ADC的IND申请进行了后续分析。他们发现,PBD-ADCs的毒性特征具有高度可比性,常见的不良事件包括血管渗漏综合征、肝酶升高、骨髓抑制、胃肠道事件、代谢效应、肌肉骨骼事件、神经病变、疼痛、呼吸困难、疲劳和肾损伤[22]。这些研究的观察结果与Zhu等人最近进行的系统评价和荟萃分析的结果一致。2000-2022年临床试验中与ADC相关的常见和严重治疗相关不良事件[23]。这些发现表明,迄今为止,大多数非现场ADC毒性与细胞毒性有效载荷的脱靶递送有关,而脱靶有效载荷递送是ADC耐受性的关键驱动因素,并最终决定了患者使用的推荐剂量。当然,在ADC的临床评估和使用过程中,靶向毒性很少成为主要问题,这并不完全出乎意料。 在临床前开发的早期阶段,可能导致严重靶向毒性的药物可能会被快速识别,从而导致在进入临床研究之前被取消选择。
从概念上讲,ADC有望通过促进细胞毒性有效载荷分子靶向递送至所需细胞群(靶向,现场毒性)来增强化疗的选择性,同时减少有效载荷向非靶向健康组织的递送,从而扩大治疗指数。在技术开发的早期阶段,抗癌ADC的预期安全性问题是靶向(即靶向介导)在具有一定程度靶抗原表达的组织中的毒性,靶标在癌细胞与健康组织中的差异表达预计将是ADC治疗指数的关键决定因素[18]。然而,随后ADC的临床经验表明,剂量限制性毒性(DLT)很少由健康组织中的靶表达驱动。在对2012年至2013年间提交的20项ADC研究性新药(IND)申请的临床前和临床数据进行详细审查时,作者发现,无论靶向抗原和抗原在健康组织中的表达程度如何,具有相同类别的接头/有效载荷的ADC通常具有高度相似的毒性特征、DLT和最大耐受剂量(MTD)[15]。例如,该综述讨论了八种正在临床开发的不同ADC,它们包含相同的vc-MMAE连接子-有效载荷组成。每个MMAE ADC都以非常相似的剂量(即在1.8 mg/kg至2.4 mg/kg的狭窄范围内)进入II期临床研究。所有 8 种 MMAE ADC 都表现出相似的 DLT(严重骨髓毒性、脓毒症和严重运动神经病变)。 所有FDA批准的vc-MMAE ADC都显示出相同的毒性特征,包括polatuzumab vedotin [ 19]、enfortumab vedotin [ 20]和tisotumab vedotin [ 21]。同样,在对2010-2014年间发表的临床ADC数据的回顾中,Masters等人发现,与ADC相关的普遍3/4级毒性与其有效载荷等级一致[12]。例如,MMAE ADC 通常报告严重贫血、中性粒细胞减少和周围神经病变。对于 DM1 ADC,通常观察到 3/4 级血小板减少症和肝毒性,而 MMAF 和 DM4 ADC 的严重眼毒性报告一致。最近,Saber和Leighton对2013年至2017年间提交的15份含有PBD-二聚体有效载荷的ADC的IND申请进行了后续分析。他们发现,PBD-ADCs的毒性特征具有高度可比性,常见的不良事件包括血管渗漏综合征、肝酶升高、骨髓抑制、胃肠道事件、代谢效应、肌肉骨骼事件、神经病变、疼痛、呼吸困难、疲劳和肾损伤[22]。这些研究的观察结果与Zhu等人最近进行的系统评价和荟萃分析的结果一致。2000-2022年临床试验中与ADC相关的常见和严重治疗相关不良事件[23]。这些发现表明,迄今为止,大多数非现场ADC毒性与细胞毒性有效载荷的脱靶递送有关,而脱靶有效载荷递送是ADC耐受性的关键驱动因素,并最终决定了患者使用的推荐剂量。当然,在ADC的临床评估和使用过程中,靶向毒性很少成为主要问题,这并不完全出乎意料。 在临床前开发的早期阶段,可能导致严重靶向毒性的药物可能会被快速识别,从而导致在进入临床研究之前被取消选择。
2.1.1. Off-Target Delivery of ADC Payloads
2.1.1. ADC有效载荷的脱靶交付
Following ADC dosing, the released (i.e., “free”) payload rapidly appears within the systemic circulation. Plasma exposure to free payload relates, in part, to premature deconjugation of the payload in the systemic circulation (e.g., due to inadequate linker stability) [24]. There are two main classes of linkers: cleavable and non-cleavable. Cleavable linkers contain chemical or enzymatic liable chemistries formulated to exploit specific conditions unique to the intracellular or tumor extracellular environments, with the goal of maintaining good stability in the systemic circulation and rapid cleavage at the target site [25,26]. In practice, cleavable linkers are often hydrolyzed in plasma at an appreciable rate, leading to the premature release of the payload in the extra-tumoral compartments. Lipophilic payloads exhibit high permeability through plasma membranes and, consequently, the released payload enters non-targeted cells efficiently (e.g., via membrane diffusion), potentially leading to unwanted cytotoxicity. For example, the hydrazone linker used in the first-generation, calicheamicin-based ADC, gemtuzumab ozogamicin, was designed to be cleaved in the acidic environment of cellular lysosomes (i.e., following cell entry via receptor-mediated endocytosis). However, this linker exhibits an appreciable rate of hydrolysis in plasma, leading to substantial payload release prior to the ADC’s engagement with the targeted antigen, and decreasing the fraction of intact ADC delivered to targeted cells [25,27].
ADC给药后,释放(即“游离”)有效载荷迅速出现在体循环中。血浆暴露于游离有效载荷在一定程度上与体循环中有效载荷的过早解偶联有关(例如,由于接头稳定性不足)[24]。接头主要有两类:可裂解和不可裂解。可切割接头含有化学或酶促化学成分,用于利用细胞内或肿瘤细胞外环境特有的特定条件,目的是在体循环中保持良好的稳定性和靶位点的快速切割[25,26]。在实践中,可切割的接头通常以可观的速率在血浆中水解,导致有效载荷在肿瘤外区室中过早释放。亲脂性有效载荷通过质膜表现出高渗透性,因此,释放的有效载荷有效地进入非靶向细胞(例如,通过膜扩散),可能导致不必要的细胞毒性。例如,第一代基于卡利霉素的ADC中使用的腙连接子吉妥珠单抗奥佐加霉素被设计为在细胞溶酶体的酸性环境中裂解(即,通过受体介导的内吞作用进入细胞后)。然而,该接头在血浆中表现出可观的水解速率,导致在ADC与靶抗原结合之前释放大量有效载荷,并降低递送至靶细胞的完整ADC的比例[25,27]。
ADC给药后,释放(即“游离”)有效载荷迅速出现在体循环中。血浆暴露于游离有效载荷在一定程度上与体循环中有效载荷的过早解偶联有关(例如,由于接头稳定性不足)[24]。接头主要有两类:可裂解和不可裂解。可切割接头含有化学或酶促化学成分,用于利用细胞内或肿瘤细胞外环境特有的特定条件,目的是在体循环中保持良好的稳定性和靶位点的快速切割[25,26]。在实践中,可切割的接头通常以可观的速率在血浆中水解,导致有效载荷在肿瘤外区室中过早释放。亲脂性有效载荷通过质膜表现出高渗透性,因此,释放的有效载荷有效地进入非靶向细胞(例如,通过膜扩散),可能导致不必要的细胞毒性。例如,第一代基于卡利霉素的ADC中使用的腙连接子吉妥珠单抗奥佐加霉素被设计为在细胞溶酶体的酸性环境中裂解(即,通过受体介导的内吞作用进入细胞后)。然而,该接头在血浆中表现出可观的水解速率,导致在ADC与靶抗原结合之前释放大量有效载荷,并降低递送至靶细胞的完整ADC的比例[25,27]。
It is important to note that ADCs with cleavable linkers may be considered to be prodrugs, and it may be expected that 100% of the administered drug (i.e., payload) is eventually liberated through linker hydrolysis. In most cases, it may be expected that the payload is eliminated from the body by a clearance (CL) process such as renal filtration, biliary excretion, or hepatic biotransformation, where the pathway of elimination and the efficiency of the free payload CL is not influenced by the site of the payload release (i.e., within plasma due to premature linker hydrolysis, or within targeted or non-targeted cells following endocytosis). The fundamental pharmacokinetic theory predicts that the cumulative exposure to the released payload in plasma (e.g., as measured by the cumulative area under the free payload plasma concentration v. time curve, AUC) is a simple function of the ADC or payload dose and the payload CL (i.e., AUC = dose/CL). As such, poor linker stability is unlikely to influence payload AUC in plasma. However, poor linker stability is expected to decrease the ratio of payload exposure in targeted sites relative to plasma, such that poor linker stability is expected to decrease the ratio of on-site to off-site ADC cytotoxicity (decreasing efficacy relative to toxicity). With the recent advancement of linker technology, several ADCs have been developed using cleavable linkers with substantially improved stability (i.e., relative to those employed in first-generation ADCs, such as gemtuzumab ozogamicin). Yet, they still face the challenge of non-selective payload deconjugation in circulation due to the susceptibility of the linkers to plasma proteases for peptide-based linkers, to plasma reactive thiols for disulfide-based and maleimide-based linkers, or to serum esterases for alkyl carbamate linkers [27,28,29,30].
需要注意的是,具有可切割接头的ADC可被视为前药,并且可以预期100%的给药药物(即有效载荷)最终通过接头水解释放。在大多数情况下,可以预期有效载荷通过清除 (CL) 过程从体内消除,例如肾脏滤过、胆汁排泄或肝脏生物转化,其中消除途径和游离有效载荷 CL 的效率不受有效载荷释放部位的影响(即,由于过早的接头水解,在血浆中, 或内吞作用后的靶向或非靶向细胞内)。基本的药代动力学理论预测,血浆中释放有效载荷的累积暴露(例如,通过自由有效载荷血浆浓度与时间曲线下的累积面积测量,AUC)是ADC或有效载荷剂量和有效载荷CL(即AUC =剂量/CL)的简单函数。因此,较差的接头稳定性不太可能影响血浆中的有效载荷AUC。然而,较差的接头稳定性预计会降低靶向位点相对于血浆的有效载荷暴露比率,因此较差的接头稳定性预计会降低现场与非现场ADC细胞毒性的比率(相对于毒性的疗效降低)。随着接头技术的最新进展,已经开发了几种使用可裂解接头的ADC,其稳定性显著提高(即,相对于第一代ADC中使用的连接子,如吉妥珠单抗奥佐霉素)。 然而,由于连接子对基于肽的连接子的血浆蛋白酶、基于二硫键和马来酰亚胺的连接子的血浆反应性硫醇或烷基氨基甲酸酯连接子的血清酯酶敏感性,它们仍然面临着循环中非选择性有效载荷解偶联的挑战[ 27, 28, 29, 30]。
需要注意的是,具有可切割接头的ADC可被视为前药,并且可以预期100%的给药药物(即有效载荷)最终通过接头水解释放。在大多数情况下,可以预期有效载荷通过清除 (CL) 过程从体内消除,例如肾脏滤过、胆汁排泄或肝脏生物转化,其中消除途径和游离有效载荷 CL 的效率不受有效载荷释放部位的影响(即,由于过早的接头水解,在血浆中, 或内吞作用后的靶向或非靶向细胞内)。基本的药代动力学理论预测,血浆中释放有效载荷的累积暴露(例如,通过自由有效载荷血浆浓度与时间曲线下的累积面积测量,AUC)是ADC或有效载荷剂量和有效载荷CL(即AUC =剂量/CL)的简单函数。因此,较差的接头稳定性不太可能影响血浆中的有效载荷AUC。然而,较差的接头稳定性预计会降低靶向位点相对于血浆的有效载荷暴露比率,因此较差的接头稳定性预计会降低现场与非现场ADC细胞毒性的比率(相对于毒性的疗效降低)。随着接头技术的最新进展,已经开发了几种使用可裂解接头的ADC,其稳定性显著提高(即,相对于第一代ADC中使用的连接子,如吉妥珠单抗奥佐霉素)。 然而,由于连接子对基于肽的连接子的血浆蛋白酶、基于二硫键和马来酰亚胺的连接子的血浆反应性硫醇或烷基氨基甲酸酯连接子的血清酯酶敏感性,它们仍然面临着循环中非选择性有效载荷解偶联的挑战[ 27, 28, 29, 30]。
In contrast to cleavable linkers, non-cleavable linkers are more stable in plasma but require the complete intracellular proteolytic catabolism of the ADCs to yield cytotoxic metabolites. These metabolites typically consist of the intact linker-payload attached to the conjugating amino acid residue from the antibodies, such as lysine-SMCC-DM1 for trastuzumab emtansine or cysteine-MC-MMAF for belantamab mafodotin [25]. These metabolites are often charged and exhibit low permeability through cell membranes [31]. Although ADCs employing non-cleavable linkers generally show a more favorable tolerability, potentially due to reduced off-target toxicity relating to free payload exposure, ADCs with cleavable linkers typically demonstrate a superior efficacy [32]. This enhanced efficacy is partially attributed to the bystander effect, which refers to the ability of the free payload to diffuse from intracellular sites of ADC catabolism and payload release to neighboring cells within the local tumor environment [33]. ADCs with lipophilic payloads and cleavable linkers, which make up more than 80% of the currently approved ADCs, are the preferred choice for the treatment of cancers associated with heterogeneous antigen expression or slow rates of antigen/ADC internalization [32]. Besides amplifying the anti-tumor potency, the bystander effect can also exacerbate the off-target toxicities of an ADC due to the increased distribution of lipophilic membrane-permeable payloads in normal tissues. For instance, in a study by Polson et al., several ADCs were constructed by conjugating antibodies against a panel of non-Hodgkin lymphoma antigens to a DM1 payload via either a cleavable (SPP) or a non-cleavable (SMCC) linker and were tested for in vivo toxicity and efficacy [34]. The SMCC-ADCs showed efficacy against only two of the seven target antigens, while the SPP-ADCs were active against all targets. However, at the dose of 20 mg ADC/kg, the animals treated with an ADC bearing the cleavable linker SPP exhibited much more significant weight loss, hepatic toxicity, and hematological toxicities when compared with the results observed following dosing with ADC employing the non-cleavable linker SMCC.
In addition to the entry of released payload into non-targeted cells via passive diffusion across plasma membranes, the non-specific endocytosis of the intact ADC may also contribute to the off-site delivery of payload. Non-specific endocytosis may be influenced by the physicochemical properties of ADCs, including hydrophobicity and charge. Since most of the drug-linker compositions utilized in ADC technology are highly lipophilic, the hydrophobicity of the ADCs is often proportional to the drug loading (i.e., drug-to-antibody ratio, DAR). In a study by Hamblett et al., several anti-CD30-vc-MMAE ADCs with DARs of two, four, and eight were evaluated for in vivo pharmacokinetics, efficacy, and toxicity [35]. It was observed that the ADCs with higher DAR values had a faster systemic clearance, a lower tolerability, and a narrower therapeutic index than the ADCs with a lower DAR. Similarly, Sun et al. demonstrated that maytansinoid-conjugated ADCs with a DAR of 10 had a 5-fold higher clearance and a decreased in vivo efficacy and tolerability compared to ADCs with a DAR lower than 6 [36]. Mice treated with low DAR ADCs (2 and 3.5) experienced less severe weight loss (approximately 4% nadir body weight loss) compared to ADCs with a DAR greater than 5.5 (7 to 9% nadir weight loss). In addition, they also observed a significantly elevated distribution of ADCs with higher DARs in the liver, possibly due to non-specific uptake by Kupffer cells and hepatic sinusoidal endothelial cells [32].
除了释放的有效载荷通过跨质膜的被动扩散进入非靶向细胞外,完整ADC的非特异性内吞作用也可能有助于有效载荷的异地递送。非特异性内吞作用可能受ADC的物理化学性质的影响,包括疏水性和电荷。由于ADC技术中使用的大多数药物连接子组合物都具有高度亲脂性,因此ADC的疏水性通常与药物负载量(即药物抗体比,DAR)成正比。在Hamblett等人的一项研究中,评估了几种DAR分别为2、4和8的抗CD30-vc-MMAE ADC的体内药代动力学、疗效和毒性[35]。结果表明,与DAR值较低的ADC相比,DAR值较高的ADC具有更快的全身清除率、更低的耐受性和更窄的治疗指数。同样,Sun等人证明,与DAR低于6的ADC相比,DAR为10的美登素偶联ADC的清除率高出5倍,体内疗效和耐受性降低[36]。与DAR大于5.5的ADC相比,用低DAR ADC(2和3.5)治疗的小鼠体重减轻(约4%的最低体重减轻)(最低体重减轻约4%)。此外,他们还观察到肝脏中DAR较高的ADC分布显著升高,这可能是由于Kupffer细胞和肝窦内皮细胞的非特异性摄取[32]。
除了释放的有效载荷通过跨质膜的被动扩散进入非靶向细胞外,完整ADC的非特异性内吞作用也可能有助于有效载荷的异地递送。非特异性内吞作用可能受ADC的物理化学性质的影响,包括疏水性和电荷。由于ADC技术中使用的大多数药物连接子组合物都具有高度亲脂性,因此ADC的疏水性通常与药物负载量(即药物抗体比,DAR)成正比。在Hamblett等人的一项研究中,评估了几种DAR分别为2、4和8的抗CD30-vc-MMAE ADC的体内药代动力学、疗效和毒性[35]。结果表明,与DAR值较低的ADC相比,DAR值较高的ADC具有更快的全身清除率、更低的耐受性和更窄的治疗指数。同样,Sun等人证明,与DAR低于6的ADC相比,DAR为10的美登素偶联ADC的清除率高出5倍,体内疗效和耐受性降低[36]。与DAR大于5.5的ADC相比,用低DAR ADC(2和3.5)治疗的小鼠体重减轻(约4%的最低体重减轻)(最低体重减轻约4%)。此外,他们还观察到肝脏中DAR较高的ADC分布显著升高,这可能是由于Kupffer细胞和肝窦内皮细胞的非特异性摄取[32]。
Positively charged molecules generally have increased charge-mediated endocytic uptake due to ionic attraction to negatively charged cell membranes [37]. Several preclinical studies have shown that the plasma clearance and tissue distribution of monoclonal immune gamma globulin (IgG) antibodies correlates with their isoelectric point (pI) [37]. For example, a study by Stuber et al. demonstrated that positively charged variants of an IgG1 antibody exhibited enhanced uptake in the liver and the spleen in mice and diminished plasma exposure compared to their neutral or negatively charged counterparts [38]. Another study by Liu et al. assessed the stability, cellular disposition, and in vivo disposition of trastuzumab (TS) mutants with incremental changes in pI from −14 to +17 [39]. They found that the positively charged variants (TS + 11, TS + 15, TS + 16, TS + 17) formed significant aggregates or failed to purify, and therefore they were excluded from subsequent experiments. In in vitro co-culture experiments with HER-2 and FcRn non-expressing Madin-Darby canine kidney cells, a significant increase in the intracellular accumulation of TS + 5 was observed compared to the negatively charged variants. Interestingly, their in vivo pharmacokinetic study indicated a U-shape relationship between charge and clearance. Highly negatively charged and positively charged variants (TS-14, TS-11, and TS + 5) displayed an elevated systemic clearance compared to variants with a more moderate negative charge (TS-8, TS-4) and the wild-type TS. Furthermore, whole-body pharmacokinetic analyses demonstrated a markedly enhanced accumulation across all major organs for TS + 5 compared to the wild-type mAb (TS) and TS-8. These observations of charge impacting the non-specific uptake and distribution of IgG might be relevant to ADCs, and the modification of the net surface charge might alter the non-specific uptake of ADCs in healthy tissues and subsequently influence their toxicity. Indeed, Zhao et al. demonstrated that altering the charge of an MMAF-conjugated ADC via the attachment of polylysine positively charged peptides or polyglutamate negatively charged peptides led to changes in the non-specific cellular uptake of the ADC in human primary corneal epithelial cells and affected its cellular cytotoxicity [40].
由于离子对带负电荷的细胞膜的吸引力,带正电荷的分子通常具有增加的电荷介导的内吞摄取[37]。几项临床前研究表明,单克隆免疫γ球蛋白(IgG)抗体的血浆清除率和组织分布与其等电点(pI)相关[37]。例如,Stuber等人的一项研究表明,与带正电荷的IgG1抗体相比,带正电荷的IgG1抗体变体在小鼠的肝脏和脾脏中表现出增强的摄取,血浆暴露减少[38]。Liu等人的另一项研究评估了曲妥珠单抗(TS)突变体的稳定性、细胞分布和体内分布,其pI从-14到+17的递增变化[39]。他们发现带正电荷的变体(TS + 11,TS + 15,TS + 16,TS + 17)形成了显着的聚集体或未能纯化,因此它们被排除在后续实验之外。在 HER-2 和 FcRn 不表达 Madin-Darby 犬肾细胞的体外共培养实验中,与带负电荷的变体相比,观察到 TS + 5 的细胞内积累显着增加。有趣的是,他们的体内药代动力学研究表明,电荷和清除率之间存在U形关系。与负电荷较中等的变体(TS-8、TS-4)和野生型 TS 相比,带高度负电荷和带正电荷的变体(TS-14、TS-11 和 TS + 5)表现出更高的全身清除率。此外,全身药代动力学分析表明,与野生型单克隆抗体 (TS) 和 TS-8 相比,TS + 5 在所有主要器官中的积累显着增强。 这些影响 IgG 非特异性摄取和分布的电荷观察结果可能与 ADC 有关,净表面电荷的改变可能会改变健康组织中 ADC 的非特异性摄取,从而影响其毒性。事实上,Zhao等人证明,通过连接带正电荷的聚赖氨酸肽或带负电荷的聚谷氨酸肽来改变MMAF偶联ADC的电荷,会导致人原代角膜上皮细胞中ADC的非特异性细胞摄取发生变化,并影响其细胞毒性[40]。
由于离子对带负电荷的细胞膜的吸引力,带正电荷的分子通常具有增加的电荷介导的内吞摄取[37]。几项临床前研究表明,单克隆免疫γ球蛋白(IgG)抗体的血浆清除率和组织分布与其等电点(pI)相关[37]。例如,Stuber等人的一项研究表明,与带正电荷的IgG1抗体相比,带正电荷的IgG1抗体变体在小鼠的肝脏和脾脏中表现出增强的摄取,血浆暴露减少[38]。Liu等人的另一项研究评估了曲妥珠单抗(TS)突变体的稳定性、细胞分布和体内分布,其pI从-14到+17的递增变化[39]。他们发现带正电荷的变体(TS + 11,TS + 15,TS + 16,TS + 17)形成了显着的聚集体或未能纯化,因此它们被排除在后续实验之外。在 HER-2 和 FcRn 不表达 Madin-Darby 犬肾细胞的体外共培养实验中,与带负电荷的变体相比,观察到 TS + 5 的细胞内积累显着增加。有趣的是,他们的体内药代动力学研究表明,电荷和清除率之间存在U形关系。与负电荷较中等的变体(TS-8、TS-4)和野生型 TS 相比,带高度负电荷和带正电荷的变体(TS-14、TS-11 和 TS + 5)表现出更高的全身清除率。此外,全身药代动力学分析表明,与野生型单克隆抗体 (TS) 和 TS-8 相比,TS + 5 在所有主要器官中的积累显着增强。 这些影响 IgG 非特异性摄取和分布的电荷观察结果可能与 ADC 有关,净表面电荷的改变可能会改变健康组织中 ADC 的非特异性摄取,从而影响其毒性。事实上,Zhao等人证明,通过连接带正电荷的聚赖氨酸肽或带负电荷的聚谷氨酸肽来改变MMAF偶联ADC的电荷,会导致人原代角膜上皮细胞中ADC的非特异性细胞摄取发生变化,并影响其细胞毒性[40]。
2.1.2. Off-Target Receptor-Mediated Uptake of ADCs
2.1.2. 脱靶受体介导的ADC摄取
As a part of the immune system, IgGs communicate with different immune cell types via the interaction of the fragment crystallizable (Fc) domain with Fc receptors expressed on the surface of the immune cells [41]. One of the major classes of Fc receptors that interact with IgG is the Fc gamma receptor (FcγR). These communications activate several IgG-mediated effector immune functions against the target; however, binding to the Fc domain may potentially lead to the target-independent uptake and toxicity of ADCs in immune cells [32]. Uppal et al. suggested that FcγRs might contribute to the frequent occurrence of thrombocytopenia associated with trastuzumab emtansine (T-DM1) treatment [42]. Thrombocytopenia was considered to be a target-independent toxicity, as platelets and platelet-forming megakaryocytes (MKs) have no expression of HER-2, the target of T-DM1 [32]. This study demonstrated that T-DM1 had minimal effects on mature MKs while being internalized and exhibiting potent cytotoxicity in differentiating MKs derived from human bone marrow. The off-target toxicity appeared to be mediated through FcγRIIa, and blocking FcγRIIa binding inhibited T-DM1 uptake. However, in another study, Zhao et al. showed that the uptake of T-DM1 in differentiating MKs was independent of FcγRIIa, but instead mediated by macropinocytosis [43]. In agreement with results from Zhao et al., a recent study by Aoyama et al. indicated that blocking FcγRIIa did not affect the cellular uptake and cytotoxicity of monomeric ADCs in MEG01-S, a FcγRIIa-expressing human megakaryoblastic leukemia cell line [44]. Instead, they observed that ADC aggregates activated FcγRs, leading to an increased uptake and cytotoxicity in FcγRs-expressing cells but not in FcγRs-negative cells. These results align with previous studies that showed enhanced activation of FcγRs by mAb aggregates, which potentially leads to the higher internalization and lysosomal degradation of mAb aggregates in immune cells compared to native mAbs [45,46]. These findings are particularly relevant for the first-generation ADCs, where aggregates commonly developed; aggregation issues have been minimized in later generation ADCs due to the optimization of several factors, including the linker/payload, DAR, and conjugation chemistries [47,48,49]. In addition to megakaryocytes, macrophages also exhibit a high expression level of FcγRs, which might render them susceptible to ADC off-target toxicity via Fc-mediated uptake [50]. Interstitial lung disease (ILD)/pneumonitis is one of the prevalent life-threatening adverse events associated with anti-HER2 ADC therapies including trastuzumab emtansine, trastuzumab deruxtecan, and trastuzumab doucamazine [51]; however, the underlying cause of ADC-induced ILD is still poorly understood. In a recently published study in monkeys, Kumagai et al. demonstrated a significant distribution of trastuzumab deruxtecan in alveolar macrophages, which localize within the alveolar space where ADC-induced pathological lesions occur [52]. Given that alveolar macrophages express high amounts of FcγR [50,53], and given that respiratory alveoli exhibit a low expression of HER2 [54], Fc-mediated non-specific uptake might contribute to ADC-induced ILD. Investigations with engineered ADCs with ablated FcγR binding might help to define the role of FcγR in ADC-induced ILD.
作为免疫系统的一部分,IgG通过片段可结晶(Fc)结构域与免疫细胞表面表达的Fc受体的相互作用与不同的免疫细胞类型进行交流[41]。与 IgG 相互作用的主要 Fc 受体之一是 Fc γ 受体 (FcγR)。这些通讯激活了几种 IgG 介导的针对靶标的效应免疫功能;然而,与Fc结构域的结合可能导致免疫细胞中ADC的靶标非依赖性摄取和毒性[32]。Uppal等人认为,FcγR可能导致曲妥珠单抗emtansine(T-DM1)治疗导致血小板减少症的频繁发生[42]。血小板减少被认为是一种靶点非依赖性毒性,因为血小板和血小板形成巨核细胞(MKs)没有表达HER-2,而HER-2是T-DM1的靶标[32]。本研究表明,T-DM1 对成熟 MK 的影响最小,同时被内化并在区分源自人骨髓的 MK 方面表现出强大的细胞毒性。脱靶毒性似乎是通过 FcγRIIa 介导的,阻断 FcγRIIa 结合抑制了 T-DM1 的摄取。然而,在另一项研究中,Zhao等人表明,T-DM1在分化MKs中的摄取与FcγRIIa无关,而是由巨胞饮作用介导的[43]。Aoyama等人最近的一项研究表明,阻断FcγRIIa不会影响表达FcγRIIa的人巨核细胞白血病细胞系MEG01-S中单体ADC的细胞摄取和细胞毒性[44]。相反,他们观察到ADC聚集体激活了FcγRs,导致表达FcγRs的细胞的摄取和细胞毒性增加,但在FcγRs阴性细胞中则没有。 这些结果与先前的研究一致,这些研究显示mAb聚集体增强了FcγR的激活,与天然mAb相比,这可能导致免疫细胞中mAb聚集体的内化和溶酶体降解更高[45,46]。这些发现与第一代ADC特别相关,因为第一代ADC通常形成聚集体;由于优化了几个因素,包括接头/有效载荷、DAR和偶联化学成分[47,48,49],在新一代ADC中,聚集问题已降至最低。除巨核细胞外,巨噬细胞还表现出高表达水平的FcγRs,这可能使它们容易通过Fc介导的摄取受到ADC脱靶毒性的影响[50]。间质性肺病(ILD)/肺炎是与抗HER2 ADC治疗相关的普遍危及生命的不良事件之一,包括曲妥珠单抗emtansine、曲妥珠单抗deruxtecan和曲妥珠单抗杜卡嗪[51];然而,ADC诱导的ILD的根本原因仍然知之甚少。在最近发表的一项猴子研究中,Kumagai等人证明了曲妥珠单抗deruxtecan在肺泡巨噬细胞中的显著分布,这些巨噬细胞位于ADC诱导的病理病变发生的肺泡腔内[52]。鉴于肺泡巨噬细胞表达大量FcγR [ 50, 53],并且呼吸道肺泡表现出低HER2表达 [ 54],Fc介导的非特异性摄取可能有助于ADC诱导的ILD。使用具有消融 FcγR 结合的工程 ADC 进行研究可能有助于确定 FcγR 在 ADC 诱导的 ILD 中的作用。
作为免疫系统的一部分,IgG通过片段可结晶(Fc)结构域与免疫细胞表面表达的Fc受体的相互作用与不同的免疫细胞类型进行交流[41]。与 IgG 相互作用的主要 Fc 受体之一是 Fc γ 受体 (FcγR)。这些通讯激活了几种 IgG 介导的针对靶标的效应免疫功能;然而,与Fc结构域的结合可能导致免疫细胞中ADC的靶标非依赖性摄取和毒性[32]。Uppal等人认为,FcγR可能导致曲妥珠单抗emtansine(T-DM1)治疗导致血小板减少症的频繁发生[42]。血小板减少被认为是一种靶点非依赖性毒性,因为血小板和血小板形成巨核细胞(MKs)没有表达HER-2,而HER-2是T-DM1的靶标[32]。本研究表明,T-DM1 对成熟 MK 的影响最小,同时被内化并在区分源自人骨髓的 MK 方面表现出强大的细胞毒性。脱靶毒性似乎是通过 FcγRIIa 介导的,阻断 FcγRIIa 结合抑制了 T-DM1 的摄取。然而,在另一项研究中,Zhao等人表明,T-DM1在分化MKs中的摄取与FcγRIIa无关,而是由巨胞饮作用介导的[43]。Aoyama等人最近的一项研究表明,阻断FcγRIIa不会影响表达FcγRIIa的人巨核细胞白血病细胞系MEG01-S中单体ADC的细胞摄取和细胞毒性[44]。相反,他们观察到ADC聚集体激活了FcγRs,导致表达FcγRs的细胞的摄取和细胞毒性增加,但在FcγRs阴性细胞中则没有。 这些结果与先前的研究一致,这些研究显示mAb聚集体增强了FcγR的激活,与天然mAb相比,这可能导致免疫细胞中mAb聚集体的内化和溶酶体降解更高[45,46]。这些发现与第一代ADC特别相关,因为第一代ADC通常形成聚集体;由于优化了几个因素,包括接头/有效载荷、DAR和偶联化学成分[47,48,49],在新一代ADC中,聚集问题已降至最低。除巨核细胞外,巨噬细胞还表现出高表达水平的FcγRs,这可能使它们容易通过Fc介导的摄取受到ADC脱靶毒性的影响[50]。间质性肺病(ILD)/肺炎是与抗HER2 ADC治疗相关的普遍危及生命的不良事件之一,包括曲妥珠单抗emtansine、曲妥珠单抗deruxtecan和曲妥珠单抗杜卡嗪[51];然而,ADC诱导的ILD的根本原因仍然知之甚少。在最近发表的一项猴子研究中,Kumagai等人证明了曲妥珠单抗deruxtecan在肺泡巨噬细胞中的显著分布,这些巨噬细胞位于ADC诱导的病理病变发生的肺泡腔内[52]。鉴于肺泡巨噬细胞表达大量FcγR [ 50, 53],并且呼吸道肺泡表现出低HER2表达 [ 54],Fc介导的非特异性摄取可能有助于ADC诱导的ILD。使用具有消融 FcγR 结合的工程 ADC 进行研究可能有助于确定 FcγR 在 ADC 诱导的 ILD 中的作用。
Besides FcγRs, mannose receptor (MR) binding and receptor-mediated internalization has been proposed as a potential mechanism that mediates the off-target hepatic toxicity of ADCs [55]. MR, which belong to the C-type lectin receptor family, are expressed widely in various tissues and play an essential role in recycling glycosylated endogenous and exogenous proteins [32]. Receptor-mediated uptake by MR is one of the critical clearance mechanisms of biotherapeutic proteins. For example, the systemic clearance of therapeutic IgGs with a high level of mannosylation is significantly faster than that of normal IgGs in humans [56]. Kogelberg et al. demonstrated MR-mediated uptake of MFECP1, a mannosylated antibody-enzyme fusion protein, by liver sinusoidal endothelial cells (SECs). Marked inhibition of MFECP1 clearance was observed with the co-administration of the MR inhibitor mannan [57]. Hepatic adverse events are frequently reported as target-independent toxicities of several ADCs [12]. In addition, acute thrombocytopenia and sinusoidal obstruction syndrome (SOS) related to ADC treatment have been linked to the degeneration and loss of liver SECs [58]. Therefore, MR-mediated uptake by liver SECs is a possible contributor to the target-independent hepatic toxicity of ADCs.
2.2. Off-Site, On-Target Toxicity
Even though evidence suggests that payload-mediated off-target mechanisms drive the majority of ADC toxicities, the binding of ADCs to target antigens expressed in healthy tissues could also lead to significant toxicities [59]. For example, around 40% of patients treated with enfortumab vedotin in clinical trials experienced dysgeusia [60], which is considered to be an on-target toxicity due to the expression of the ADC target (nectin-4) in the salivary glands [61]. Indeed, this toxicity is uncommon in patients treated with other approved MMAE-ADCs, including brentuximab vedotin, polatuzumab vedotin, and tisotumab vedotin [62,63,64]. As shown in the example above, ADCs producing toxicity that is not typically associated with their payload may be suggestive of an on-target mechanism. The converse is also true; the observation of a common toxicity for several ADCs targeting the same antigen with different payloads is also suggestive of an on-target mechanism. For instance, ILD and pneumonitis were identified as adverse events leading to the dose modification, dose delay, or treatment discontinuation of the anti-HER2 ADC trastuzumab deruxtecan in the phase 2 DESTINY-Breast01 clinical trial [65]. These same toxicities, severe and lethal cases of ILD and pneumonitis, also occurred in patients treated with additional anti-HER2 ADCs, including trastuzumab duocarmazine and trastuzumab emtansine [66,67]. In addition, serious cardiac toxicity, including a decrease in left ventricular ejection fraction (LVEF), has been observed in patients treated with the trastuzumab-based ADCs mentioned above. These toxicities are also included in the black box warning for trastuzumab treatment [55], suggesting that on-target uptake of the ADC, rather than the non-specific, off-target uptake of payload, is the primary driver of toxicity.
尽管有证据表明,有效载荷介导的脱靶机制驱动了大多数ADC毒性,但ADC与健康组织中表达的靶抗原的结合也可能导致显著的毒性[59]。例如,在临床试验中,约40%接受enfortumab vedotin治疗的患者出现味觉障碍[ 60],这被认为是由于唾液腺中ADC靶标(nectin-4)的表达而引起的靶向毒性[ 61]。事实上,这种毒性在接受其他已获批的MMAE-ADC治疗的患者中并不常见,包括brentuximab vedotin、polatuzumab vedotin和tisotumab vedotin[62,63,64]。如上例所示,产生的毒性通常与其有效载荷无关的ADC可能提示靶向机制。反之亦然;对靶向相同抗原且具有不同有效载荷的几种ADC的共同毒性的观察也表明了靶向机制。例如,在2期DESTINY-Breast01临床试验中,ILD和肺炎被确定为导致抗HER2 ADC曲妥珠单抗deruxtecan剂量调整、剂量延迟或治疗停止的不良事件[65]。这些相同的毒性,严重和致命的ILD和肺炎病例,也发生在接受其他抗HER2 ADC治疗的患者中,包括曲妥珠单抗、多卡玛嗪和曲妥珠单抗emtansine[66,67]。此外,在接受上述基于曲妥珠单抗的 ADC 治疗的患者中观察到严重的心脏毒性,包括左心室射血分数 (LVEF) 降低。 这些毒性也包含在曲妥珠单抗治疗的黑匣子警告中[ 55],表明ADC的靶向摄取,而不是有效载荷的非特异性脱靶摄取,是毒性的主要驱动因素。
尽管有证据表明,有效载荷介导的脱靶机制驱动了大多数ADC毒性,但ADC与健康组织中表达的靶抗原的结合也可能导致显著的毒性[59]。例如,在临床试验中,约40%接受enfortumab vedotin治疗的患者出现味觉障碍[ 60],这被认为是由于唾液腺中ADC靶标(nectin-4)的表达而引起的靶向毒性[ 61]。事实上,这种毒性在接受其他已获批的MMAE-ADC治疗的患者中并不常见,包括brentuximab vedotin、polatuzumab vedotin和tisotumab vedotin[62,63,64]。如上例所示,产生的毒性通常与其有效载荷无关的ADC可能提示靶向机制。反之亦然;对靶向相同抗原且具有不同有效载荷的几种ADC的共同毒性的观察也表明了靶向机制。例如,在2期DESTINY-Breast01临床试验中,ILD和肺炎被确定为导致抗HER2 ADC曲妥珠单抗deruxtecan剂量调整、剂量延迟或治疗停止的不良事件[65]。这些相同的毒性,严重和致命的ILD和肺炎病例,也发生在接受其他抗HER2 ADC治疗的患者中,包括曲妥珠单抗、多卡玛嗪和曲妥珠单抗emtansine[66,67]。此外,在接受上述基于曲妥珠单抗的 ADC 治疗的患者中观察到严重的心脏毒性,包括左心室射血分数 (LVEF) 降低。 这些毒性也包含在曲妥珠单抗治疗的黑匣子警告中[ 55],表明ADC的靶向摄取,而不是有效载荷的非特异性脱靶摄取,是毒性的主要驱动因素。
Interestingly, the application of the same ADC to treat different cancers may lead to different toxicities [8]. For example, severe rash was one of the dose-limiting toxicities of glembatumumab vedotin, which might be an on-target toxicity relating to the expression of its target, gpNMB, in the skin [68,69]. Following the administration of the same dose of glembatumumab vedotin, grade ≥ 3 rash occurred in 4% of patients with advanced breast cancer but in 30% of patients with advanced myeloma [69,70]. Pruritus and alopecia also occurred more frequently in melanoma patients than in breast cancer patients (63% and 65% vs. 21% and 25%, respectively). The mechanism behind this phenomenon is still unknown, but it is possibly related to the effects of the two types of cancer on the expression of gpNMB by healthy cells. In another example, LOP628, an anti-KIT-SMCC-DM1 ADC, caused life-threatening rapid hypersensitivity reactions (HSR) in several patients, which led to the termination of its clinical development [71]. Data from L’Italien et al. implicate that mast cell degranulation due to co-engagement of KIT and FcγRs by the parental anti-KIT mAb is the leading cause of HSR [71]. This particular case presents a unique mechanism in which the on-target toxicity does not involve cytotoxicity to the antigen-expressing cells but rather works by activating the immune system through a costimulatory signaling pathway. Additionally, the target antigen expression in healthy tissues does not always lead to on-target toxicity. For instance, the expression of the membrane-associated mucin MUC16 was reported in the human ocular surface epithelia; nevertheless, no ocular toxicity occurred in patients treated with DMUC5754A, an anti-MUC16 MMAE-ADC, in clinical trials [72,73]. Another antigen, TROP-2, is known to be expressed widely in various tissues [74]; however, the clinical toxicity profile of sacituzumab govitecan, an anti-TROP2 SN-38 conjugated ADC, largely resembles that of the SN-38 payload, suggestive of off-target mechanisms [59]. The low on-target toxicity of sacituzumab govitecan might relate to: (1) the limited accessibility of the antigen in non-malignant tissues compared to tumors; (2) an insufficient expression level to induce toxicities; and (3) the lower sensitivity of normal tissues to the SN-38 payload compared to cancerous tissues [75]. In addition, since sacituzumab govitecan employs a pH-sensitive linker engineered to release the payload within an acidic environment, the higher pH found within the extracellular fluid of normal tissues relative to tumors might contribute to the low on-target toxicity observed for this ADC [76].
有趣的是,应用相同的ADC治疗不同的癌症可能会导致不同的毒性[8]。例如,严重皮疹是glembatumumab vedotin的剂量限制性毒性之一,这可能是与其靶标gpNMB在皮肤中的表达有关的靶向毒性[68,69]。在给予相同剂量的glembatumumab vedotin后,4%的晚期乳腺癌患者发生≥级3级皮疹,30%的晚期骨髓瘤患者发生3级皮疹[69,70]。瘙痒和脱发在黑色素瘤患者中的发生率也高于乳腺癌患者(分别为 63% 和 65% 对 21% 和 25%)。这种现象背后的机制尚不清楚,但可能与两种癌症对健康细胞表达gpNMB的影响有关。在另一个例子中,LOP628是一种抗KIT-SMCC-DM1 ADC,在几名患者中引起危及生命的快速超敏反应(rapid hypersensitivity reaction, HSR),导致其临床开发终止[71]。L'Italien等人的数据表明,由于亲本抗KIT单克隆抗体共结合KIT和FcγR导致的肥大细胞脱颗粒是HSR的主要原因[71]。这种特殊情况呈现出一种独特的机制,其中靶向毒性不涉及对抗原表达细胞的细胞毒性,而是通过共刺激信号通路激活免疫系统起作用。此外,健康组织中的靶抗原表达并不总是导致靶向毒性。例如,膜相关粘蛋白MUC16在人眼表上皮细胞中的表达被报道;然而,在临床试验中,接受抗MUC16 MMAE-ADC DMUC5754A治疗的患者没有发生眼毒性[72,73]。 已知另一种抗原TROP-2在各种组织中广泛表达[74];然而,抗TROP2 SN-38偶联ADC的临床毒性特征与SN-38有效载荷的临床毒性特征基本相似,提示脱靶机制[59]。sacituzumab govitecan的低靶向毒性可能与以下原因有关:(1)与肿瘤相比,抗原在非恶性组织中的可及性有限;(2)表达水平不足以诱发毒性;(3)与癌组织相比,正常组织对SN-38有效载荷的敏感性较低[75]。此外,由于sacituzumab govitecan采用pH敏感接头,旨在在酸性环境中释放有效载荷,因此在正常组织的细胞外液中发现的相对于肿瘤的pH值较高,可能有助于观察到该ADC的低靶向毒性[ 76]。
有趣的是,应用相同的ADC治疗不同的癌症可能会导致不同的毒性[8]。例如,严重皮疹是glembatumumab vedotin的剂量限制性毒性之一,这可能是与其靶标gpNMB在皮肤中的表达有关的靶向毒性[68,69]。在给予相同剂量的glembatumumab vedotin后,4%的晚期乳腺癌患者发生≥级3级皮疹,30%的晚期骨髓瘤患者发生3级皮疹[69,70]。瘙痒和脱发在黑色素瘤患者中的发生率也高于乳腺癌患者(分别为 63% 和 65% 对 21% 和 25%)。这种现象背后的机制尚不清楚,但可能与两种癌症对健康细胞表达gpNMB的影响有关。在另一个例子中,LOP628是一种抗KIT-SMCC-DM1 ADC,在几名患者中引起危及生命的快速超敏反应(rapid hypersensitivity reaction, HSR),导致其临床开发终止[71]。L'Italien等人的数据表明,由于亲本抗KIT单克隆抗体共结合KIT和FcγR导致的肥大细胞脱颗粒是HSR的主要原因[71]。这种特殊情况呈现出一种独特的机制,其中靶向毒性不涉及对抗原表达细胞的细胞毒性,而是通过共刺激信号通路激活免疫系统起作用。此外,健康组织中的靶抗原表达并不总是导致靶向毒性。例如,膜相关粘蛋白MUC16在人眼表上皮细胞中的表达被报道;然而,在临床试验中,接受抗MUC16 MMAE-ADC DMUC5754A治疗的患者没有发生眼毒性[72,73]。 已知另一种抗原TROP-2在各种组织中广泛表达[74];然而,抗TROP2 SN-38偶联ADC的临床毒性特征与SN-38有效载荷的临床毒性特征基本相似,提示脱靶机制[59]。sacituzumab govitecan的低靶向毒性可能与以下原因有关:(1)与肿瘤相比,抗原在非恶性组织中的可及性有限;(2)表达水平不足以诱发毒性;(3)与癌组织相比,正常组织对SN-38有效载荷的敏感性较低[75]。此外,由于sacituzumab govitecan采用pH敏感接头,旨在在酸性环境中释放有效载荷,因此在正常组织的细胞外液中发现的相对于肿瘤的pH值较高,可能有助于观察到该ADC的低靶向毒性[ 76]。
3. Clinical Toxicity Profiles of Antibody-Drug Conjugates
第3章 抗体-药物偶联物的临床毒性特征
3.1. Approved ADCs 3.1. 批准的ADC
In this section, the clinical toxicity profiles of the approved ADCs are discussed in detail. A summary of these information can be found in Table 1.
在本节中,将详细讨论已批准的ADC的临床毒性特征。表 1 中提供了这些信息的摘要。
在本节中,将详细讨论已批准的ADC的临床毒性特征。表 1 中提供了这些信息的摘要。

3.1.1. ADCs with Calicheamicin Payload
3.1.1. 具有卡利霉素有效载荷的ADC
Gemtuzumab Ozogamicin (Mylotarg)
吉妥珠单抗奥佐霉素(Mylotarg)
Gemtuzumab ozogamicin consists of a humanized anti-CD33 IgG linked to a DNA-alkylating calicheamicin payload via a pH-sensitive hydrazone linker [77,78]. In the first phase 1 dose-escalation study, 40 patients with relapsed or refractory CD33-positive acute myeloid leukemia (AML) were treated with 0.25 mg/m2 to 9 mg/m2 of gemtuzumab ozogamicin [79]. The dose of 9 mg/m2 was selected for the subsequent phase 2 study because saturation of the CD33 binding sites, regardless of the leukemic burden, was observed at this dose level. In the phase 2 study, 142 patients with first relapse CD33-positive AML were treated with 9 mg/m2 of gemtuzumab ozogamicin once every two weeks [80]. The most common adverse events of all grades (≥30%) included thrombocytopenia, fatigue, neutropenia, pyrexia, nausea, infection, chills, hemorrhage, vomiting, headache, stomatitis, diarrhea, and abdominal pain. Most of the treated patients experienced grade 3 or 4 neutropenia (97%) and thrombocytopenia (99%). Other grade ≥3 treatment-related adverse events included increases in AST or ALT levels (17%), sepsis (16%), fever (15%), chills (13%), nausea and vomiting (11%), dyspnea (9%), hypertension (9%), hypotension (8%), pneumonia (7%), and asthenia (7%). The clinically relevant serious adverse events were neutropenia (34.3%), thrombocytopenia (21.7%), and infusion-related reactions (2.5%). The most common causes of treatment discontinuation were infection, hemorrhage, multi-organ failure, and veno-occlusive disease/sinusoidal obstruction syndrome (VOD/SOS).
吉妥珠单抗奥佐米星由人源化抗CD33 IgG组成,通过pH敏感的腙接头与DNA烷基化卡利霉素有效载荷连接[77,78]。在第一个1期剂量递增研究中,40例复发或难治性CD33阳性急性髓系白血病(AML)患者接受了0.25mg/m-9mg 2 /m 2 的吉妥珠单抗奥佐米星治疗[79]。在随后的 2 期研究中选择了 9 mg/m 2 的剂量,因为无论白血病负荷如何,在该剂量水平下都观察到 CD33 结合位点的饱和度。在2期研究中,142例首次复发CD33阳性AML患者每两周接受一次9mg/m 2 的吉妥珠单抗奥佐米星治疗[80]。所有级别最常见的不良事件(≥30%)包括血小板减少、疲劳、中性粒细胞减少、发热、恶心、感染、寒战、出血、呕吐、头痛、口腔炎、腹泻和腹痛。大多数接受治疗的患者出现 3 级或 4 级中性粒细胞减少症 (97%) 和血小板减少症 (99%)。其他 ≥3 级治疗相关不良事件包括 AST 或 ALT 水平升高 (17%)、脓毒症 (16%)、发烧 (15%)、寒战 (13%)、恶心和呕吐 (11%)、呼吸困难 (9%)、高血压 (9%)、低血压 (8%)、肺炎 (7%) 和虚弱 (7%)。临床相关严重不良事件为中性粒细胞减少(34.3%)、血小板减少(21.7%)和输液相关反应(2.5%)。中止治疗的最常见原因是感染、出血、多器官衰竭和静脉闭塞性疾病/窦阻塞综合征 (VOD/SOS)。
吉妥珠单抗奥佐米星由人源化抗CD33 IgG组成,通过pH敏感的腙接头与DNA烷基化卡利霉素有效载荷连接[77,78]。在第一个1期剂量递增研究中,40例复发或难治性CD33阳性急性髓系白血病(AML)患者接受了0.25mg/m-9mg 2 /m 2 的吉妥珠单抗奥佐米星治疗[79]。在随后的 2 期研究中选择了 9 mg/m 2 的剂量,因为无论白血病负荷如何,在该剂量水平下都观察到 CD33 结合位点的饱和度。在2期研究中,142例首次复发CD33阳性AML患者每两周接受一次9mg/m 2 的吉妥珠单抗奥佐米星治疗[80]。所有级别最常见的不良事件(≥30%)包括血小板减少、疲劳、中性粒细胞减少、发热、恶心、感染、寒战、出血、呕吐、头痛、口腔炎、腹泻和腹痛。大多数接受治疗的患者出现 3 级或 4 级中性粒细胞减少症 (97%) 和血小板减少症 (99%)。其他 ≥3 级治疗相关不良事件包括 AST 或 ALT 水平升高 (17%)、脓毒症 (16%)、发烧 (15%)、寒战 (13%)、恶心和呕吐 (11%)、呼吸困难 (9%)、高血压 (9%)、低血压 (8%)、肺炎 (7%) 和虚弱 (7%)。临床相关严重不良事件为中性粒细胞减少(34.3%)、血小板减少(21.7%)和输液相关反应(2.5%)。中止治疗的最常见原因是感染、出血、多器官衰竭和静脉闭塞性疾病/窦阻塞综合征 (VOD/SOS)。
Gemtuzumab ozogamicin was approved in 2001 by the FDA; however, post-marketing studies revealed significant systemic toxicities and poor efficacy [81]. In phase 3 randomized comparative trial SWOGS0106, 637 AML patients were treated with either gemtuzumab ozogamicin in combination with other chemotherapeutic agents, including daunorubicin and cytosine arabinoside, or chemotherapy alone [82]. While the combination therapy with gemtuzumab ozogamicin showed no significant clinical benefit, a marked increase in the mortality rate due to toxicity was observed in the combination arm (5.7%) compared to the chemotherapy arm (1.4%). Based on these results, gemtuzumab ozogamicin was voluntarily withdrawn in June 2010. Subsequently, several clinical studies at lower recommended doses and a different dosing schedule demonstrated improved clinical outcomes and more favorable toxicity profiles, which led to the re-approval of gemtuzumab ozogamicin in 2017 [83,84,85].
吉妥珠单抗奥佐米星于2001年获得FDA批准;然而,上市后研究显示其全身毒性显著,疗效较差[81]。在3期随机比较试验SWOGS0106中,637例AML患者接受吉妥珠单抗奥佐米星联合其他化疗药物(包括柔红霉素和胞嘧啶阿拉伯糖苷)或单独化疗[82]。虽然吉妥珠单抗奥佐米星的联合治疗没有显示出明显的临床益处,但与化疗组(1.4%)相比,联合治疗组(5.7%)的毒性死亡率显着增加。基于这些结果,吉妥珠单抗奥佐米星于2010年6月自愿停药。随后,几项较低推荐剂量和不同给药方案的临床研究显示,临床结局有所改善,毒性特征更有利,这导致吉妥珠单抗奥佐米星在2017年再次获批[83,84,85]。
吉妥珠单抗奥佐米星于2001年获得FDA批准;然而,上市后研究显示其全身毒性显著,疗效较差[81]。在3期随机比较试验SWOGS0106中,637例AML患者接受吉妥珠单抗奥佐米星联合其他化疗药物(包括柔红霉素和胞嘧啶阿拉伯糖苷)或单独化疗[82]。虽然吉妥珠单抗奥佐米星的联合治疗没有显示出明显的临床益处,但与化疗组(1.4%)相比,联合治疗组(5.7%)的毒性死亡率显着增加。基于这些结果,吉妥珠单抗奥佐米星于2010年6月自愿停药。随后,几项较低推荐剂量和不同给药方案的临床研究显示,临床结局有所改善,毒性特征更有利,这导致吉妥珠单抗奥佐米星在2017年再次获批[83,84,85]。
Hepatotoxicity, including life-threatening and sometimes fatal hepatic VOD events in patients receiving gemtuzumab ozogamicin as a single agent or as part of a combination chemotherapy regimen, is included in the black box warning [86]. In phase 2 clinical trials conducted in 277 AML patients, VOD occurred in 5% of the patients and caused fatal reactions in 3% [87,88]. The expression of CD33 in hepatocytes is likely the leading cause of gemtuzumab ozogamicin-mediated hepatotoxicity [89,90]. In addition, clinical trials of the anti-CD33 ADC vadastuximab talirine (SGN-CD33A) were put on hold in December 2016 due to the induction of VOD/SOS in clinical settings which resulted in the death of four out of six SOS cases [91]. Besides hepatocytes, CD33 is also highly expressed in hematopoietic cells, leading to the significant hematologic toxicities of anti-CD33 therapies [92,93].
肝毒性,包括吉妥珠单抗奥佐米星单药或联合化疗方案一部分患者的肝毒性,包括危及生命、有时甚至致命的肝脏VOD事件,包含在黑匣子警告中[ 86]。在277例AML患者中进行的2期临床试验中,VOD发生率为5%,引起致命反应的患者为3%[87,88]。CD33在肝细胞中的表达可能是吉妥珠单抗奥佐霉素介导的肝毒性的主要原因[89,90]。此外,2016年12月,抗CD33 ADC瓦达妥昔单抗塔利林(sadustumimab talirine, SGN-CD33A)的临床试验被搁置,原因是在临床环境中诱导VOD/SOS,导致6例SOS病例中有4例死亡[91]。除肝细胞外,CD33在造血细胞中也高度表达,导致抗CD33疗法具有显著的血液学毒性[92,93]。
肝毒性,包括吉妥珠单抗奥佐米星单药或联合化疗方案一部分患者的肝毒性,包括危及生命、有时甚至致命的肝脏VOD事件,包含在黑匣子警告中[ 86]。在277例AML患者中进行的2期临床试验中,VOD发生率为5%,引起致命反应的患者为3%[87,88]。CD33在肝细胞中的表达可能是吉妥珠单抗奥佐霉素介导的肝毒性的主要原因[89,90]。此外,2016年12月,抗CD33 ADC瓦达妥昔单抗塔利林(sadustumimab talirine, SGN-CD33A)的临床试验被搁置,原因是在临床环境中诱导VOD/SOS,导致6例SOS病例中有4例死亡[91]。除肝细胞外,CD33在造血细胞中也高度表达,导致抗CD33疗法具有显著的血液学毒性[92,93]。
Inotuzumab Ozogamicin (Besponsa)
Inotuzumab Ozogamicin (Besponsa)
Inotuzumab ozogamicin consists of a humanized anti-CD22 IgG linked to a calicheamicin payload via a pH-sensitive hydrazone linker [94]. In a phase 1/2 dose-escalation and dose-expansion study, relapsed/refractory acute lymphoblastic leukemia patients were treated with inotuzumab ozogamicin at doses of 1.2 mg/kg to 1.8 mg/kg per 28-day cycle [95]. Neutropenia and thrombocytopenia were the most common treatment-related adverse events. There were four reported cases of VOD/SOS, including one fatal case. The dose of 1.8 mg/m2 (0.8 mg/m2 on day 1; 0.5 mg/m2 on days 8 and 15) was selected for the subsequent phase 3 clinical trial.
Inotuzumab ozogamicin 由人源化抗 CD22 IgG 组成,通过 pH 敏感的腙接头与卡利霉素有效载荷连接 [ 94]。一项1/2期剂量递增和剂量扩展研究显示,复发/难治性急性淋巴细胞白血病患者接受inotuzumab ozogamicin治疗,剂量为1.2mg/kg-1.8mg/kg/2日[95]。中性粒细胞减少和血小板减少是最常见的治疗相关不良事件。报告了4例VOD/SOS病例,包括1例死亡病例。1.8 mg/m 2 (第 1 天 0.8 mg/m 2 ;第 8 天和第 15 天 0.5 mg/m 2 )的剂量被选为随后的 3 期临床试验。
Inotuzumab ozogamicin 由人源化抗 CD22 IgG 组成,通过 pH 敏感的腙接头与卡利霉素有效载荷连接 [ 94]。一项1/2期剂量递增和剂量扩展研究显示,复发/难治性急性淋巴细胞白血病患者接受inotuzumab ozogamicin治疗,剂量为1.2mg/kg-1.8mg/kg/2日[95]。中性粒细胞减少和血小板减少是最常见的治疗相关不良事件。报告了4例VOD/SOS病例,包括1例死亡病例。1.8 mg/m 2 (第 1 天 0.8 mg/m 2 ;第 8 天和第 15 天 0.5 mg/m 2 )的剂量被选为随后的 3 期临床试验。
In a phase 3 randomized open-label INO-VATE ALL study, patients with relapsed/refractory acute lymphoblastic leukemia were treated with either 1.8 mg/m2 of inotuzumab ozogamicin per cycle (n = 139) or standard chemotherapy (n = 120) [96]. Among the patients treated with inotuzumab ozogamicin, the most common (≥15%) treatment-related adverse events of all grades were neutropenia (36%), thrombocytopenia (29%), infection (48%), anemia (18%), leukopenia (17%), febrile neutropenia (16%), and nausea (15%). The grade ≥3 treatment-related adverse events most commonly reported (≥10%) were neutropenia (34%), thrombocytopenia (20%), leukopenia (15%), febrile neutropenia (14%), anemia (11%), and lymphopenia (11%). Fatal infections, including pneumonia, neutropenic sepsis, sepsis, septic shock, and pseudomonal sepsis, were reported in 5% of patients. Dose reductions and discontinuation due to adverse events occurred in 2% and 9% of patients. Cases of VOD/SOS occurred in 14% of patients, including five fatal cases (3%) [97]. Patients who underwent hematopoietic stem cell transplants after inotuzumab ozogamicin treatment have an increased risk of VOD. As a result, hepatotoxicity is included in the black box warning for patients who received inotuzumab ozogamicin [98]. Since CD22 is not expressed in the liver, the target-independent uptake mechanisms of either the ADC or the free payload play a potential role in hepatotoxicity [91].
一项3期随机开放标签INO-VATE ALL研究显示,复发/难治性急性淋巴细胞白血病患者每周期接受1.8mg/m 2 的inotuzumab ozogamicin(n=139)或标准化疗(n=120)[96]。在接受inotuzumab奥佐霉素治疗的患者中,所有级别中最常见的(≥15%)治疗相关不良事件是中性粒细胞减少(36%)、血小板减少(29%)、感染(48%)、贫血(18%)、白细胞减少(17%)、发热性中性粒细胞减少(16%)和恶心(15%)。最常报告的 ≥3 级治疗相关不良事件 (≥10%) 是中性粒细胞减少症 (34%)、血小板减少症 (20%)、白细胞减少症 (15%)、发热性中性粒细胞减少症 (14%)、贫血 (11%) 和淋巴细胞减少症 (11%)。5% 的患者报告了致命性感染,包括肺炎、中性粒细胞减少性脓毒症、脓毒症、脓毒性休克和假单胞菌性脓毒症。2%和9%的患者因不良事件而减少剂量和停药。14%的患者发生VOD/SOS病例,包括5例死亡病例(3%)[97]。在inotuzumab ozogamicin治疗后接受造血干细胞移植的患者发生VOD的风险增加。因此,肝毒性被纳入接受inotuzumab ozogamicin治疗的患者的黑匣子警告中[ 98]。由于CD22在肝脏中不表达,ADC或游离有效载荷的靶标非依赖性摄取机制在肝毒性中起潜在作用[91]。
一项3期随机开放标签INO-VATE ALL研究显示,复发/难治性急性淋巴细胞白血病患者每周期接受1.8mg/m 2 的inotuzumab ozogamicin(n=139)或标准化疗(n=120)[96]。在接受inotuzumab奥佐霉素治疗的患者中,所有级别中最常见的(≥15%)治疗相关不良事件是中性粒细胞减少(36%)、血小板减少(29%)、感染(48%)、贫血(18%)、白细胞减少(17%)、发热性中性粒细胞减少(16%)和恶心(15%)。最常报告的 ≥3 级治疗相关不良事件 (≥10%) 是中性粒细胞减少症 (34%)、血小板减少症 (20%)、白细胞减少症 (15%)、发热性中性粒细胞减少症 (14%)、贫血 (11%) 和淋巴细胞减少症 (11%)。5% 的患者报告了致命性感染,包括肺炎、中性粒细胞减少性脓毒症、脓毒症、脓毒性休克和假单胞菌性脓毒症。2%和9%的患者因不良事件而减少剂量和停药。14%的患者发生VOD/SOS病例,包括5例死亡病例(3%)[97]。在inotuzumab ozogamicin治疗后接受造血干细胞移植的患者发生VOD的风险增加。因此,肝毒性被纳入接受inotuzumab ozogamicin治疗的患者的黑匣子警告中[ 98]。由于CD22在肝脏中不表达,ADC或游离有效载荷的靶标非依赖性摄取机制在肝毒性中起潜在作用[91]。
3.1.2. ADCs with Auristatin Payloads
3.1.2. 具有 Auristatin 有效载荷的 ADC
Brentuximab Vedotin (Adcetris)
Brentuximab Vedotin(Adcetris)
Brentuximab vedotin is a CD30-targeting chimeric IgG1 conjugated to MMAE via a protease cleavable valine-citrulline (vc) linker [99]. The first phase I clinical study of this ADC was conducted in 45 relapsed or refractory patients with CD30-positive hematologic malignancies, including Hodgkin’s lymphoma (HL), anaplastic large-cell lymphoma (ALCL), and angioimmunoblastic T-cell lymphoma [100]. A traditional 3 + 3 dose-escalation study design was performed with intravenous infusion of brentuximab vedotin at doses of 0.1 to 3.6 mg/kg every three weeks as salvage therapy. A single patient who received the 3.6 mg/kg dose experienced febrile neutropenia leading to sepsis and death two weeks after dosing. Dose-limiting toxicities were observed in several patients treated with the 2.7 mg/kg dose, including grade 3 hyperglycemia, unrelated grade 3 acute renal failure, and unrelated grade 3 prostatitis and febrile neutropenia. Only one patient in the 1.8 mg/kg dose cohort experienced dose-limiting toxicity (grade 4 thrombocytopenia). Based on this observation, the dose of 1.8 mg/kg was identified as the maximum tolerated dose. The most common adverse effects observed in this trial were peripheral neuropathy, neutropenia, pyrexia, diarrhea, and nausea.
Brentuximab vedotin 是一种靶向 CD30 的嵌合 IgG1,通过蛋白酶可裂解的缬氨酸-瓜氨酸 (vc) 接头与 MMAE 偶联 [ 99]。该ADC的首个I期临床研究纳入了45例CD30阳性血液系统恶性肿瘤复发或难治性患者,包括霍奇金淋巴瘤(HL)、间变性大细胞淋巴瘤(ALCL)和血管免疫母细胞性T细胞淋巴瘤[100]。传统的 3 + 3 剂量递增研究设计进行,每三周静脉输注 0.1 至 3.6 mg/kg 的 brentuximab vedotin 作为挽救治疗。一名接受 3.6 mg/kg 剂量的患者在给药两周后出现发热性中性粒细胞减少症,导致败血症和死亡。在接受 2.7 mg/kg 剂量治疗的几名患者中观察到剂量限制性毒性,包括 3 级高血糖、无关的 3 级急性肾功能衰竭以及无关的 3 级前列腺炎和发热性中性粒细胞减少症。在 1.8 mg/kg 剂量队列中,只有 1 例患者出现剂量限制性毒性(4 级血小板减少症)。根据这一观察结果,1.8 mg/kg的剂量被确定为最大耐受剂量。该试验中观察到的最常见的不良反应是周围神经病变、中性粒细胞减少、发热、腹泻和恶心。
Brentuximab vedotin 是一种靶向 CD30 的嵌合 IgG1,通过蛋白酶可裂解的缬氨酸-瓜氨酸 (vc) 接头与 MMAE 偶联 [ 99]。该ADC的首个I期临床研究纳入了45例CD30阳性血液系统恶性肿瘤复发或难治性患者,包括霍奇金淋巴瘤(HL)、间变性大细胞淋巴瘤(ALCL)和血管免疫母细胞性T细胞淋巴瘤[100]。传统的 3 + 3 剂量递增研究设计进行,每三周静脉输注 0.1 至 3.6 mg/kg 的 brentuximab vedotin 作为挽救治疗。一名接受 3.6 mg/kg 剂量的患者在给药两周后出现发热性中性粒细胞减少症,导致败血症和死亡。在接受 2.7 mg/kg 剂量治疗的几名患者中观察到剂量限制性毒性,包括 3 级高血糖、无关的 3 级急性肾功能衰竭以及无关的 3 级前列腺炎和发热性中性粒细胞减少症。在 1.8 mg/kg 剂量队列中,只有 1 例患者出现剂量限制性毒性(4 级血小板减少症)。根据这一观察结果,1.8 mg/kg的剂量被确定为最大耐受剂量。该试验中观察到的最常见的不良反应是周围神经病变、中性粒细胞减少、发热、腹泻和恶心。
In the pivotal phase II trial, 102 patients with relapsed or refractory HL after autologous stem cell transplantation were treated with 1.8 mg/kg of brentuximab vedotin by IV infusion every three weeks as salvage therapy [101]. The most common therapeutic-related adverse events of any grade were peripheral sensory neuropathy (42%), nausea (35%), fatigue (34%), neutropenia (19%), diarrhea (18%), pyrexia (14%), vomiting (13%), arthralgia (12%), pruritus (12%), myalgia (11%), peripheral motor neuropathy (11%), and alopecia (10%). Serious adverse events of grade 3 and 4 severity were observed in 55% of patients, including neutropenia (20%), peripheral sensory neuropathy (8%), thrombocytopenia (8%), and anemia (6%). Treatment discontinuation due to adverse events occurred in 20% of patients, with peripheral sensory neuropathy (6%) and peripheral motor neuropathy (3%) being the most common events. Dose delays occurred in 8% of patients, with neutropenia (16%) and peripheral sensory neuropathy (13%) being the most common causes. Dose reductions from 1.8 to 1.2 mg/kg were required in 11 patients, mostly due to peripheral neuropathy (10 of 11 patients). A similar toxicity profile was observed in another phase II clinical trial for patients with relapsed or refractory systemic anaplastic large-cell lymphoma, with additional common grade 1 or 2 adverse effects including rash (24%), constipation (22%), headache (19%), cough (17%), dyspnea (17%), upper respiratory tract infection (17%), decreased appetite (16%), dizziness (16%), insomnia (16%), chills (14%), muscle spasms (14%), thrombocytopenia (14%), weight loss (14%), edema peripheral (12%), and pain in extremity (12%) [102].
在一项关键的II期试验中,102例自体干细胞移植后复发或难治性霍奇金淋巴瘤患者每3周静脉输注1.8mg/kg的brentuximab vedotin作为挽救性治疗[101]。任何级别最常见的治疗相关不良事件是周围感觉神经病变 (42%)、恶心 (35%)、疲劳 (34%)、中性粒细胞减少 (19%)、腹泻 (18%)、发热 (14%)、呕吐 (13%)、关节痛 (12%)、瘙痒 (12%)、肌痛 (11%)、周围运动神经病变 (11%) 和脱发 (10%)。55%的患者出现严重不良事件,包括中性粒细胞减少(20%)、周围感觉神经病变(8%)、血小板减少(8%)和贫血(6%)。20% 的患者因不良事件而停止治疗,其中周围感觉神经病变 (6%) 和周围运动神经病变 (3%) 是最常见的事件。8%的患者出现剂量延迟,中性粒细胞减少(16%)和周围感觉神经病变(13%)是最常见的原因。11 例患者需要将剂量从 1.8 mg/kg 减少到 1.2 mg/kg,主要是由于周围神经病变(11 例患者中有 10 例)。在另一项针对复发或难治性系统性间变性大细胞淋巴瘤患者的 II 期临床试验中观察到类似的毒性特征,并伴有其他常见的 1 级或 2 级不良反应,包括皮疹 (24%)、便秘 (22%)、头痛 (19%)、咳嗽 (17%)、呼吸困难 (17%)、上呼吸道感染 (17%)、食欲下降 (16%)、头晕 (16%)、失眠 (16%)、发冷 (14%)、 肌肉痉挛(14%)、血小板减少(14%)、体重减轻(14%)、外周水肿(12%)和四肢疼痛(12%)[102]。
在一项关键的II期试验中,102例自体干细胞移植后复发或难治性霍奇金淋巴瘤患者每3周静脉输注1.8mg/kg的brentuximab vedotin作为挽救性治疗[101]。任何级别最常见的治疗相关不良事件是周围感觉神经病变 (42%)、恶心 (35%)、疲劳 (34%)、中性粒细胞减少 (19%)、腹泻 (18%)、发热 (14%)、呕吐 (13%)、关节痛 (12%)、瘙痒 (12%)、肌痛 (11%)、周围运动神经病变 (11%) 和脱发 (10%)。55%的患者出现严重不良事件,包括中性粒细胞减少(20%)、周围感觉神经病变(8%)、血小板减少(8%)和贫血(6%)。20% 的患者因不良事件而停止治疗,其中周围感觉神经病变 (6%) 和周围运动神经病变 (3%) 是最常见的事件。8%的患者出现剂量延迟,中性粒细胞减少(16%)和周围感觉神经病变(13%)是最常见的原因。11 例患者需要将剂量从 1.8 mg/kg 减少到 1.2 mg/kg,主要是由于周围神经病变(11 例患者中有 10 例)。在另一项针对复发或难治性系统性间变性大细胞淋巴瘤患者的 II 期临床试验中观察到类似的毒性特征,并伴有其他常见的 1 级或 2 级不良反应,包括皮疹 (24%)、便秘 (22%)、头痛 (19%)、咳嗽 (17%)、呼吸困难 (17%)、上呼吸道感染 (17%)、食欲下降 (16%)、头晕 (16%)、失眠 (16%)、发冷 (14%)、 肌肉痉挛(14%)、血小板减少(14%)、体重减轻(14%)、外周水肿(12%)和四肢疼痛(12%)[102]。
In the AETHERA study, a randomized, double-blind, placebo-controlled phase III clinical trial, patients with Hodgkin’s lymphoma who were at risk of relapse or progression after autologous stem cell transplantation were treated with brentuximab vedotin as consolidation therapy [103]. The most common adverse events at any grade occurring in the brentuximab vedotin-treated group at a significantly higher incidence compared to the placebo group were peripheral sensory neuropathy (56% vs. 16%), neutropenia (35% vs. 12%), and peripheral motor neuropathy (23% vs. 2%). Severe adverse events at grade ≥3 also included peripheral sensory neuropathy (10% vs. 1%), neutropenia (29% vs. 10%), and peripheral motor neuropathy (6% vs. 1%). The incidences of treatment discontinuation and dose modifications (dose reduction or delay) due to peripheral neuropathy were 23% and 31%, respectively. Neutropenia caused dose delays in 22% of patients.
AETHERA研究是一项随机、双盲、安慰剂对照的III期临床试验,对自体干细胞移植后有复发或进展风险的霍奇金淋巴瘤患者接受brentuximab vedotin治疗[103]。与安慰剂组相比,brentuximab vedotin治疗组发生的任何级别最常见的不良事件是周围感觉神经病变(56% vs. 16%)、中性粒细胞减少(35% vs. 12%)和周围运动神经病变(23% vs. 2%)。≥3级严重不良事件还包括周围感觉神经病变(10% vs. 1%)、中性粒细胞减少(29% vs. 10%)和周围运动神经病变(6% vs. 1%)。周围神经病变导致的治疗中断和剂量调整(剂量减少或延迟)的发生率分别为 23% 和 31%。中性粒细胞减少导致 22% 的患者出现剂量延迟。
AETHERA研究是一项随机、双盲、安慰剂对照的III期临床试验,对自体干细胞移植后有复发或进展风险的霍奇金淋巴瘤患者接受brentuximab vedotin治疗[103]。与安慰剂组相比,brentuximab vedotin治疗组发生的任何级别最常见的不良事件是周围感觉神经病变(56% vs. 16%)、中性粒细胞减少(35% vs. 12%)和周围运动神经病变(23% vs. 2%)。≥3级严重不良事件还包括周围感觉神经病变(10% vs. 1%)、中性粒细胞减少(29% vs. 10%)和周围运动神经病变(6% vs. 1%)。周围神经病变导致的治疗中断和剂量调整(剂量减少或延迟)的发生率分别为 23% 和 31%。中性粒细胞减少导致 22% 的患者出现剂量延迟。
During post-marketing surveillance, several fatal cases of progressive multifocal leukoencephalopathy resulting from John Cunningham virus infection have been reported. Therefore, the black box warning for brentuximab vedotin has included this potential risk [62]. Other severe and fatal adverse events associated with brentuximab vedotin include febrile neutropenia, hepatotoxicity, pneumonitis, interstitial lung disease, acute respiratory distress syndrome, Stevens-Johnson syndrome, toxic epidermal necrolysis, acute pancreatitis, perforation, hemorrhage, erosion, ulcer, intestinal obstruction, enterocolitis, neutropenic colitis, and ileus.
在上市后监测期间,已报告了几例由约翰·坎宁安病毒感染引起的进行性多灶性脑白质病的致命病例。因此,brentuximab vedotin的黑匣子警告包括了这种潜在风险[ 62]。与brentuximab vedotin相关的其他严重和致死性不良事件包括发热性中性粒细胞减少症、肝毒性、肺炎、间质性肺病、急性呼吸窘迫综合征、Stevens-Johnson综合征、中毒性表皮坏死松解症、急性胰腺炎、穿孔、出血、糜烂、溃疡、肠梗阻、小肠结肠炎、中性粒细胞减少性结肠炎和肠梗阻。
在上市后监测期间,已报告了几例由约翰·坎宁安病毒感染引起的进行性多灶性脑白质病的致命病例。因此,brentuximab vedotin的黑匣子警告包括了这种潜在风险[ 62]。与brentuximab vedotin相关的其他严重和致死性不良事件包括发热性中性粒细胞减少症、肝毒性、肺炎、间质性肺病、急性呼吸窘迫综合征、Stevens-Johnson综合征、中毒性表皮坏死松解症、急性胰腺炎、穿孔、出血、糜烂、溃疡、肠梗阻、小肠结肠炎、中性粒细胞减少性结肠炎和肠梗阻。
Polatuzumab Vedotin (Polivy)
Polatuzumab Vedotin (Polivy)
Polatuzumab Vedotin consists of a humanized anti-CD79b IgG linked to an MMAE payload via a protease cleavable valine-citrulline linker [104]. The first-in-human phase I clinical trial of polatuzumab vedotin was conducted in two dose-escalation cohorts, relapsed/refractory non-Hodgkin’s lymphoma (NHL) patients (n = 34) and chronic lymphocytic leukemia (CLL) patients (n = 18) [19]. In the NHL cohort, patients were treated with polatuzumab vedotin at 0.1 to 2.4 mg/kg doses every three weeks. Dose-limiting toxicity (grade 4 neutropenia) was observed in one of the ten patients (10%) treated at a 2.4 mg/kg dose, the recommended dose for the subsequent phase 2 study. In the CLL cohort, patients were treated with polatuzumab vedotin at 0.25 to 1.8 mg/kg doses. Dose-limiting adverse events (grade 4 neutropenia and grade 4 fungal infection) occurred in two of the five patients (40%) treated at a 1.8 mg/kg dose. None of the 18 patients with CLL achieved objective responses. Another two dose-expansion NHL cohorts received either 2.4 mg/kg polatuzumab vedotin alone (n = 45) or in combination with rituximab (n = 9). Among the 45 NHL patients treated with polatuzumab vedotin at the recommended phase 2 dose of 2.4 mg/kg, 26 (58%) experienced at least one grade 3–4 adverse event, with the most common adverse events reported in more than two patients including neutropenia (40%), anemia (11%), and peripheral neuropathy (9%). Treatment discontinuation due to adverse events occurred in 23 (51%) patients, with peripheral sensory neuropathy being the most common cause (11 of 45 patients). A treatment delay of at least one dose occurred in 17 (38%) patients, with neutropenia being the most common reason (11 of 45 patients). A dose reduction to 1.8 mg/kg occurred in six (13%) patients due to neutropenia (two patients), sensory neuropathy (two patients), paranesthesia (one patient), and diarrhea (one patient). Among the nine patients with NHL treated with the combination therapy of polatuzumab vedotin and rituximab, grade 3–4 adverse events occurred in seven patients (77%), with neutropenia (five patients), anemia (two patients), and febrile neutropenia (two patients) being the most common.
Polatuzumab Vedotin 由一种人源化抗 CD79b IgG 组成,通过蛋白酶可裂解的缬氨酸-瓜氨酸接头与 MMAE 有效载荷相连 [ 104]。polatuzumab vedotin的首次人体I期临床试验在两个剂量递增队列中进行,即复发/难治性非霍奇金淋巴瘤(NHL)患者(n=34)和慢性淋巴细胞白血病(CLL)患者(n=18)[19]。在 NHL 队列中,患者每三周接受 0.1 至 2.4 mg/kg 剂量的 polatuzumab vedotin 治疗。在以 2.4 mg/kg 剂量治疗的 10 名患者中,有 1 名 (10%) 观察到剂量限制性毒性(4 级中性粒细胞减少症),这是随后 2 期研究的推荐剂量。在CLL队列中,患者接受0.25至1.8mg/kg剂量的polatuzumab vedotin治疗。剂量限制性不良事件(4 级中性粒细胞减少症和 4 级真菌感染)发生在以 1.8 mg/kg 剂量治疗的 5 例患者中,有 2 例 (40%)发生。18例CLL患者均未达到客观缓解。另外两个剂量扩展 NHL 队列单独接受 2.4 mg/kg polatuzumab vedotin (n = 45) 或与利妥昔单抗联合 (n = 9)。在推荐的 2 期剂量为 2.4 mg/kg 的 45 名 NHL 患者中,26 名 (58%) 至少经历了一次 3-4 级不良事件,最常见的不良事件发生在两名以上的患者中,包括中性粒细胞减少 (40%)、贫血 (11%) 和周围神经病变 (9%)。23例(51%)患者因不良事件而停止治疗,周围感觉神经病变是最常见的原因(45例患者中有11例)。17 例 (38%) 患者出现至少一剂治疗延迟,中性粒细胞减少是最常见的原因(45 例患者中有 11 例)。剂量减少到 1.6 例 (13%) 患者因中性粒细胞减少(2 例)、感觉神经病变(2 例)、异常性呼吸(1 例)和腹泻(1 例)发生 8 mg/kg。在接受polatuzumab vedotin和利妥昔单抗联合治疗的9例NHL患者中,7例(77%)发生3-4级不良事件,其中中性粒细胞减少(5例)、贫血(2例)和发热性中性粒细胞减少(2例)最常见。
Polatuzumab Vedotin 由一种人源化抗 CD79b IgG 组成,通过蛋白酶可裂解的缬氨酸-瓜氨酸接头与 MMAE 有效载荷相连 [ 104]。polatuzumab vedotin的首次人体I期临床试验在两个剂量递增队列中进行,即复发/难治性非霍奇金淋巴瘤(NHL)患者(n=34)和慢性淋巴细胞白血病(CLL)患者(n=18)[19]。在 NHL 队列中,患者每三周接受 0.1 至 2.4 mg/kg 剂量的 polatuzumab vedotin 治疗。在以 2.4 mg/kg 剂量治疗的 10 名患者中,有 1 名 (10%) 观察到剂量限制性毒性(4 级中性粒细胞减少症),这是随后 2 期研究的推荐剂量。在CLL队列中,患者接受0.25至1.8mg/kg剂量的polatuzumab vedotin治疗。剂量限制性不良事件(4 级中性粒细胞减少症和 4 级真菌感染)发生在以 1.8 mg/kg 剂量治疗的 5 例患者中,有 2 例 (40%)发生。18例CLL患者均未达到客观缓解。另外两个剂量扩展 NHL 队列单独接受 2.4 mg/kg polatuzumab vedotin (n = 45) 或与利妥昔单抗联合 (n = 9)。在推荐的 2 期剂量为 2.4 mg/kg 的 45 名 NHL 患者中,26 名 (58%) 至少经历了一次 3-4 级不良事件,最常见的不良事件发生在两名以上的患者中,包括中性粒细胞减少 (40%)、贫血 (11%) 和周围神经病变 (9%)。23例(51%)患者因不良事件而停止治疗,周围感觉神经病变是最常见的原因(45例患者中有11例)。17 例 (38%) 患者出现至少一剂治疗延迟,中性粒细胞减少是最常见的原因(45 例患者中有 11 例)。剂量减少到 1.6 例 (13%) 患者因中性粒细胞减少(2 例)、感觉神经病变(2 例)、异常性呼吸(1 例)和腹泻(1 例)发生 8 mg/kg。在接受polatuzumab vedotin和利妥昔单抗联合治疗的9例NHL患者中,7例(77%)发生3-4级不良事件,其中中性粒细胞减少(5例)、贫血(2例)和发热性中性粒细胞减少(2例)最常见。
In the ROMULUS phase 2 clinical trial, patients with relapsed/refractory diffuse large B-cell lymphoma (DLBCL, n = 81) or follicular lymphoma (FL, n = 41) were randomized to receive 375 mg/m2 of rituximab plus 2.4 mg/kg of either polatuzumab vedotin (R-pola) or pinatuzumab vedotin (R-pina) [105]. Pinatuzumab vedotin is an investigational anti-CD22-MMAE ADC also developed by Genentech. The most common treatment-related adverse events reported for both treatments were diarrhea, fatigue, peripheral neuropathy, nausea, and neutropenia. Among patients treated with R-pola, the most common grade 3–4 adverse events in the DLBCL cohort were neutropenia (23%), anemia (8%), and diarrhea (8%), and those in the FL cohort were neutropenia (15%) and diarrhea (10%). Peripheral neuropathy occurred in more than half of the patients treated with R-pola and resulted in treatment discontinuations in 18% and 55% of the patients in the DLBCL and FL cohorts, respectively. Consequently, the maximal dose of polatuzumab vedotin in combination therapy used for the subsequent dose-escalation clinical trial was reduced to 1.8 mg/kg.
The pivotal GO29365 phase 1b/2 clinical trial was conducted to evaluate the safety and efficacy of the combination treatments of polatuzumab vedotin (1.8 mg/kg) with bedamustine plus either obinutuzumab (pola-BG) or rituximab (pola-BR) in transplantation-ineligible relapsed/refractory DLBCL [106]. An initial safety phase 1b included six patients treated with pola-BR and six patients treated with pola-BG, followed by a phase 2 including an expansion cohort treated with pola-BG (n = 21) and a cohort (n = 80) randomly assigned to treatments of either pola-BR or BR alone. Among the cohort with treatment randomized to BR or pola-BR, the grade 3–4 adverse events that were more common with the pola-BR treatment were anemia (28% vs. 18%), thrombocytopenia (41% vs. 23%), and neutropenia (46% vs. 33%). Peripheral neutropenia and diarrhea of all grades were more prevalent with pola-BR (44% and 39% vs. 3% and 28%, respectively). A polatuzumab vedotin dose reduction occurred in 5% of the patient solely due to peripheral neuropathy, and treatment discontinuation due to adverse events was more common with the pola-BR treatment (33% vs. 10%). Similar results were observed in another clinical trial in the Japanese population [107].
Enfortumab Vedotin (Padcev)
Enfortumab vedotin is composed of a fully human anti-nectin-4 IgG conjugated to an MMAE payload via a protease cleavable valine-citrulline linker [61]. The phase 1 EV-101 dose-escalation/dose-expansion study was conducted in patients with nectin-4-positive solid tumors [20]. This study consisted of three parts: (A) to establish the maximum tolerated dose and the recommended phase 2 dose of enfortumab vedotin; (B) to evaluate enfortumab vedotin in three dose-expansion cohorts, including patients with metastatic urothelial carcinomas (mUC) and severe renal insufficiency, patients with non-small-cell lung cancer, and patients with ovarian cancer; (C) to evaluate enfortumab vedotin in a dose-expansion cohort in mUC patients previously treated with an immune checkpoint inhibitor. The recommended phase 2 dose was determined to be 1.25 mg/kg. Among the 155 mUC patients, the most common adverse events of any grade were fatigue (53%), alopecia (46%), decreased appetite (42%), dysgeusia (38%), nausea (38%), peripheral sensory neuropathy (38%), pruritus (35%), diarrhea (33%), and maculopapular rash (27%). A similar toxicity profile was observed in the EV-201 phase II clinical trial, which consisted of a single-arm study of enfortumab vedotin 1.25 mg/kg in 125 patients with advanced or metastatic UC who were previously treated with platinum chemotherapy or immune checkpoint inhibitors [108]. Treatment-related grade 3–4 adverse events occurred in 54% of the patients, with rash (17%), neutropenia (8%), anemia (7%), and fatigue (6%) being the most common.
In the two-arm phase 3 EV-301 clinical trial, patients with advanced or metastatic UC who had previously received platinum chemotherapy or immune checkpoint inhibitors were treated with either enfortumab vedotin (1.25 mg/kg, n = 296) or chemotherapy (n = 291) [109]. In the cohort treated with enfortumab vedotin, the most common grade ≥3 adverse events (≥5%) were maculopapular rash (7.4%), fatigue (6.4%), and decreased neutrophil count (6.1%). Dose delays occurred in 61% of patients, with peripheral neuropathy (23%), rash (11%), and fatigue (9%) being the most common reasons (≥4%). Dose reductions occurred in 34% of patients, with peripheral neuropathy (10%), rash (8%), decreased appetite (3%), and fatigue (3%) being the most common reasons (≥2%). Treatment discontinuation occurred in 17% of patients, with peripheral neuropathy (5%) and rash (4%) being the most common reasons (≥2%).
在一项双臂3期EV-301临床试验中,既往接受过铂类化疗或免疫检查点抑制剂的晚期或转移性溃疡性结肠炎患者接受enfortumab vedotin(1.25mg/kg,n=296)或化疗(n=291)治疗[109]。在接受 enfortumab vedotin 治疗的队列中,最常见的 ≥3 级不良事件 (≥5%) 是斑丘疹 (7.4%)、疲劳 (6.4%) 和中性粒细胞计数减少 (6.1%)。61%的患者出现剂量延迟,最常见的原因为周围神经病变(23%)、皮疹(11%)和疲劳(9%)(≥4%)。34%的患者出现剂量减少,最常见的原因是周围神经病变(10%)、皮疹(8%)、食欲下降(3%)和疲劳(3%)(≥2%)。17%的患者停止治疗,周围神经病变(5%)和皮疹(4%)是最常见的原因(≥2%)。
在一项双臂3期EV-301临床试验中,既往接受过铂类化疗或免疫检查点抑制剂的晚期或转移性溃疡性结肠炎患者接受enfortumab vedotin(1.25mg/kg,n=296)或化疗(n=291)治疗[109]。在接受 enfortumab vedotin 治疗的队列中,最常见的 ≥3 级不良事件 (≥5%) 是斑丘疹 (7.4%)、疲劳 (6.4%) 和中性粒细胞计数减少 (6.1%)。61%的患者出现剂量延迟,最常见的原因为周围神经病变(23%)、皮疹(11%)和疲劳(9%)(≥4%)。34%的患者出现剂量减少,最常见的原因是周围神经病变(10%)、皮疹(8%)、食欲下降(3%)和疲劳(3%)(≥2%)。17%的患者停止治疗,周围神经病变(5%)和皮疹(4%)是最常见的原因(≥2%)。
Severe skin reactions are included in the black box warning for enfortumab vedotin [60]. Treatment-related skin reactions, which occurred in 55% of the patients treated with enfortumab vedotin in clinical trials, were expected as on-target off-tumor toxicities due to nectin-4 expression in the skin [110]. Grade ≥3 skin reactions occurred in 13% of patients, leading to dose interruption/reduction in 9% of patients and treatment discontinuation in 2.6% of patients [60]. Severe cutaneous adverse reactions, including fatal Stevens-Johnson syndrome and toxic epidermal necrolysis, were reported during clinical trials and post-marketing settings [110]. Skin adverse reactions were also common in patients treated with other ADCs utilizing an MMAE payload, including brentuximab vedotin (31%), glembatumumab vedotin (44%), and polatuzumab vedotin (13–31%), suggesting a potential contribution of MMAE to skin toxicities [62,63,111,112]. Hyperglycemia and pneumonitis are also included in the warning box due to several life-threatening or fatal reactions observed during clinical trials [60]. In clinical trials, hyperglycemia of any grade occurred in 14% of patients treated with enfortumab vedotin, with 7% of patients developed grade 3–4 hyperglycemia. Discontinuation of treatment due to hyperglycemia occurred in 0.6% of patients. Pneumonitis of any grade occurred in 3.1% of the patients, and discontinuation of the treatment due to grade 3–4 pneumonitis occurred in 0.7% of the patients.
严重的皮肤反应包含在enfortumab vedotin的黑匣子警告中[ 60]。在临床试验中,55%接受enfortumab vedotin治疗的患者发生治疗相关皮肤反应,预计由于皮肤中nectin-4表达,靶向脱瘤毒性[110]。13%的患者发生≥3级皮肤反应,导致9%的患者剂量中断/减少,2.6%的患者停止治疗[60]。在临床试验和上市后环境中报告了严重的皮肤不良反应,包括致命的Stevens-Johnson综合征和中毒性表皮坏死松解症[110]。皮肤不良反应在使用MMAE有效载荷的其他ADC治疗的患者中也很常见,包括brentuximab vedotin(31%)、glembatumumab vedotin(44%)和polatuzumab vedotin(13-31%),表明MMAE对皮肤毒性的潜在贡献[62,63,111,112]。高血糖和肺炎也被列入警告框,因为在临床试验中观察到一些危及生命或致命的反应[60]。在临床试验中,接受 enfortumab vedotin 治疗的患者中有 14% 发生任何级别的高血糖,其中 7% 的患者出现 3-4 级高血糖。0.6%的患者因高血糖而停止治疗。3.1%的患者发生任何级别的肺炎,0.7%的患者因3-4级肺炎而停止治疗。
严重的皮肤反应包含在enfortumab vedotin的黑匣子警告中[ 60]。在临床试验中,55%接受enfortumab vedotin治疗的患者发生治疗相关皮肤反应,预计由于皮肤中nectin-4表达,靶向脱瘤毒性[110]。13%的患者发生≥3级皮肤反应,导致9%的患者剂量中断/减少,2.6%的患者停止治疗[60]。在临床试验和上市后环境中报告了严重的皮肤不良反应,包括致命的Stevens-Johnson综合征和中毒性表皮坏死松解症[110]。皮肤不良反应在使用MMAE有效载荷的其他ADC治疗的患者中也很常见,包括brentuximab vedotin(31%)、glembatumumab vedotin(44%)和polatuzumab vedotin(13-31%),表明MMAE对皮肤毒性的潜在贡献[62,63,111,112]。高血糖和肺炎也被列入警告框,因为在临床试验中观察到一些危及生命或致命的反应[60]。在临床试验中,接受 enfortumab vedotin 治疗的患者中有 14% 发生任何级别的高血糖,其中 7% 的患者出现 3-4 级高血糖。0.6%的患者因高血糖而停止治疗。3.1%的患者发生任何级别的肺炎,0.7%的患者因3-4级肺炎而停止治疗。
Tisotumab Vedotin (Tivdak)
Tisotumab Vedotin (Tivdak)
Tisotumab vedotin is a conjugate of a human anti-tissue factor monoclonal antibody and an MMAE payload via a protease cleavable valine-citruline linker [113]. In the dose-escalation phase of the first-in-human phase 1/2 InnovaTV 201 study, 27 patients with a diverse array of advanced or metastatic solid tumors, including ovary, cervix, endometrium, bladder, prostate, esophagus, non-small-cell lung cancer, or squamous cell carcinoma of the head and neck, were treated with 0.3 mg/kg to 2.2 mg/kg of tisotumab vedotin once every three weeks [21]. Three patients in the 2.2 mg/kg dose cohort had dose-limiting toxicities, including type 2 diabetes mellitus, mucositis, and neutropenic fever. Therefore, the maximum tolerated dose and recommended phase 2 dose was determined as 2.0 mg/kg. In the dose-expansion phase of this study, 147 patients with cancer of the ovary, cervix, endometrium, bladder, prostate, and esophagus, as well as non-small-cell lung cancer, were treated with 2.0 mg/kg of tisotumab vedotin once every three weeks. Across tumor types in phase 2, the most common (≥20%) treatment-related adverse events of any grade were epistaxis (69%), fatigue (56%), nausea (52%), alopecia (44%), conjunctivitis (43%), decreased appetite (36%), constipation (35%), diarrhea (30%), vomiting (29%), peripheral neuropathy (22%), dry eye (22%), and abdominal pain (20%). The most common (>2%) treatment-related adverse events of grade 3 or worse were fatigue (10%), anemia (5%), abdominal pain (4%), hypokalemia (4%), conjunctivitis (3%), hyponatremia (3%), and vomiting (3%).
Tisotumab vedotin 是人抗组织因子单克隆抗体和 MMAE 有效载荷的偶联物,通过蛋白酶可裂解的缬氨酸-瓜氨酸连接子 [ 113]。在首次人体1/2期InnovaTV 201研究的剂量递增阶段,27例患有卵巢、宫颈、子宫内膜、膀胱、前列腺、食管、非小细胞肺癌或头颈部鳞状细胞癌等多种晚期或转移性实体瘤的患者每3周接受一次0.3mg/kg-2.2mg/kg的tisotumab vedotin治疗[21]。2.2 mg/kg 剂量队列中有 3 名患者出现剂量限制性毒性,包括 2 型糖尿病、粘膜炎和中性粒细胞减少性发热。因此,最大耐受剂量和推荐的 2 期剂量确定为 2.0 mg/kg。在这项研究的剂量扩展阶段,147 名患有卵巢癌、宫颈癌、子宫内膜癌、膀胱癌、前列腺癌和食道癌以及非小细胞肺癌的患者每三周接受一次 2.0 mg/kg 的 tisotumab vedotin 治疗。在2期肿瘤类型中,最常见(≥20%)的治疗相关不良事件是鼻衄(69%)、疲劳(56%)、恶心(52%)、脱发(44%)、结膜炎(43%)、食欲下降(36%)、便秘(35%)、腹泻(30%)、呕吐(29%)、周围神经病变(22%)、干眼症(22%)和腹痛(20%)。最常见的(>2%)3级或更差的治疗相关不良事件是疲劳(10%)、贫血(5%)、腹痛(4%)、低钾血症(4%)、结膜炎(3%)、低钠血症(3%)和呕吐(3%)。
Tisotumab vedotin 是人抗组织因子单克隆抗体和 MMAE 有效载荷的偶联物,通过蛋白酶可裂解的缬氨酸-瓜氨酸连接子 [ 113]。在首次人体1/2期InnovaTV 201研究的剂量递增阶段,27例患有卵巢、宫颈、子宫内膜、膀胱、前列腺、食管、非小细胞肺癌或头颈部鳞状细胞癌等多种晚期或转移性实体瘤的患者每3周接受一次0.3mg/kg-2.2mg/kg的tisotumab vedotin治疗[21]。2.2 mg/kg 剂量队列中有 3 名患者出现剂量限制性毒性,包括 2 型糖尿病、粘膜炎和中性粒细胞减少性发热。因此,最大耐受剂量和推荐的 2 期剂量确定为 2.0 mg/kg。在这项研究的剂量扩展阶段,147 名患有卵巢癌、宫颈癌、子宫内膜癌、膀胱癌、前列腺癌和食道癌以及非小细胞肺癌的患者每三周接受一次 2.0 mg/kg 的 tisotumab vedotin 治疗。在2期肿瘤类型中,最常见(≥20%)的治疗相关不良事件是鼻衄(69%)、疲劳(56%)、恶心(52%)、脱发(44%)、结膜炎(43%)、食欲下降(36%)、便秘(35%)、腹泻(30%)、呕吐(29%)、周围神经病变(22%)、干眼症(22%)和腹痛(20%)。最常见的(>2%)3级或更差的治疗相关不良事件是疲劳(10%)、贫血(5%)、腹痛(4%)、低钾血症(4%)、结膜炎(3%)、低钠血症(3%)和呕吐(3%)。
In the phase 2 open-label single-arm InnovaTV204 study, 101 patients with recurrent or metastatic cervical cancer with disease progression on or after chemotherapy were treated with 2.0 mg/kg of tisotumab vedotin every three weeks until disease progression or unacceptable toxicity [114]. The most common (≥25%) adverse reactions, including laboratory abnormalities, were decreased hemoglobin (59%), fatigue (57%), a decrease in lymphocytes (50%), nausea (41%), peripheral neuropathy (46%), alopecia ((39%), epistaxis (39%), conjunctival adverse reactions (37%), hemorrhage (38%), a decrease in leukocytes (30%), increased creatinine, dry eye (29%), an increased prothrombin international normalized ratio (26%), a prolonged activated partial thromboplastin time (26%), diarrhea (27%), and rash (25%) [64]. Serious adverse reactions occurred in 43% of patients, with ileus (6%), hemorrhage (5%), pneumonia (4%), peripheral neuropathy (3%), sepsis (3%), constipation (3%), and pyrexia (3%) being the most common. A dose interruption occurred in 47% of patients, with peripheral neuropathy (8%), conjunctival adverse reactions (4%), and hemorrhage (4%) being the most common reasons. A dose reduction occurred in 23% of patients, with conjunctival adverse reactions (9%) and corneal adverse reactions (8%) being the most common reasons. Treatment discontinuation occurred in 13% of patients, with peripheral neuropathy (5%) and corneal adverse reactions (4%) being the most common reasons.
在2期开放标签单臂InnovaTV204研究中,101例化疗期间或化疗后疾病进展的复发或转移性宫颈癌患者每3周接受2.0mg/kg替索妥单抗vedotin治疗,直至疾病进展或出现不可接受的毒性[114]。最常见的(≥25%)不良反应,包括实验室异常,是血红蛋白降低(59%)、疲劳(57%)、淋巴细胞减少(50%)、恶心(41%)、周围神经病变(46%)、脱发((39%)、鼻出血(39%)、结膜不良反应(37%)、出血(38%)、白细胞减少(30%)、肌酐升高、干眼症(29%)、凝血酶原国际标准化比值升高(26%)、活化部分凝血活酶时间延长(26%)、 腹泻(27%)和皮疹(25%)[64]。43%的患者发生严重不良反应,最常见的是肠梗阻(6%)、出血(5%)、肺炎(4%)、周围神经病变(3%)、脓毒症(3%)、便秘(3%)和发热(3%)。47%的患者发生剂量中断,最常见的原因是周围神经病变(8%)、结膜不良反应(4%)和出血(4%)。23%的患者出现剂量减少,结膜不良反应(9%)和角膜不良反应(8%)是最常见的原因。13%的患者停止治疗,最常见的原因是周围神经病变(5%)和角膜不良反应(4%)。
在2期开放标签单臂InnovaTV204研究中,101例化疗期间或化疗后疾病进展的复发或转移性宫颈癌患者每3周接受2.0mg/kg替索妥单抗vedotin治疗,直至疾病进展或出现不可接受的毒性[114]。最常见的(≥25%)不良反应,包括实验室异常,是血红蛋白降低(59%)、疲劳(57%)、淋巴细胞减少(50%)、恶心(41%)、周围神经病变(46%)、脱发((39%)、鼻出血(39%)、结膜不良反应(37%)、出血(38%)、白细胞减少(30%)、肌酐升高、干眼症(29%)、凝血酶原国际标准化比值升高(26%)、活化部分凝血活酶时间延长(26%)、 腹泻(27%)和皮疹(25%)[64]。43%的患者发生严重不良反应,最常见的是肠梗阻(6%)、出血(5%)、肺炎(4%)、周围神经病变(3%)、脓毒症(3%)、便秘(3%)和发热(3%)。47%的患者发生剂量中断,最常见的原因是周围神经病变(8%)、结膜不良反应(4%)和出血(4%)。23%的患者出现剂量减少,结膜不良反应(9%)和角膜不良反应(8%)是最常见的原因。13%的患者停止治疗,最常见的原因是周围神经病变(5%)和角膜不良反应(4%)。
Ocular toxicity is included in the black box warning for patients treated with tisotumab vedotin [64]. Immunohistochemical evaluation in normal human ocular tissues confirmed the expression of tissue factor in the ocular epithelium, suggesting a target-mediated uptake of tisotumab vedotin in the eye is the likely cause of toxicity [115,116]. Ocular adverse reactions occurred in 60% of patients with cervical cancer across clinical trials, with conjunctival adverse reactions (40%), dry eye (29%), corneal adverse reactions (21%), and blepharitis (8%) being the most common. Grade 3 ocular adverse reactions occurred in 3.8% of patients, including severe ulcerative keratitis in 3.2% of patients. Ocular adverse reactions led to the discontinuation of tisotumab vedotin in 6% of patients with cervical cancer.
对于接受tisotumab vedotin治疗的患者,眼部毒性包含在黑匣子警告中[ 64]。正常人眼组织的免疫组织化学评估证实了组织因子在眼上皮中的表达,表明靶向介导的 tisotumab vedotin 在眼睛中的摄取是毒性的可能原因 [ 115, 116]。在临床试验中,60%的宫颈癌患者发生眼部不良反应,其中结膜不良反应(40%)、干眼症(29%)、角膜不良反应(21%)和睑缘炎(8%)最为常见。3.8%的患者发生3级眼部不良反应,包括3.2%的患者出现严重溃疡性角膜炎。眼部不良反应导致6%的宫颈癌患者停用tisotumab vedotin。
对于接受tisotumab vedotin治疗的患者,眼部毒性包含在黑匣子警告中[ 64]。正常人眼组织的免疫组织化学评估证实了组织因子在眼上皮中的表达,表明靶向介导的 tisotumab vedotin 在眼睛中的摄取是毒性的可能原因 [ 115, 116]。在临床试验中,60%的宫颈癌患者发生眼部不良反应,其中结膜不良反应(40%)、干眼症(29%)、角膜不良反应(21%)和睑缘炎(8%)最为常见。3.8%的患者发生3级眼部不良反应,包括3.2%的患者出现严重溃疡性角膜炎。眼部不良反应导致6%的宫颈癌患者停用tisotumab vedotin。
Belantamab Mafodotin (Blenrep)
Belantamab Mafodotin (Blenrep)
Belantamab mafodotin consists of a humanized afucosylated Fc-engineered IgG targeting B-cell maturation antigen (BCMA) covalently linked to MMAF payload via a non-cleavable maleimidocaproyl (mc) linker. The first-in-human phase 1 DREAMM-1 clinical trial explored belantamab mafodotin as monotherapy in relapsed or refractory multiple myeloma (RRMM) [117]. This study comprised two parts: (I) a dose-escalation phase assessing the safety and tolerability of belantamab mafodotin and determining the recommended phase 2 dose; (II) a dose-expansion phase evaluating the safety and tolerability pharmacokinetics and clinical activity of the recommended phase 2 dose. In part I, 38 patients received 0.03 to 4.6 mg/kg belantamab mafodotin every three weeks. The treatment was well-tolerated with no maximal tolerated dose established. The recommended phase 2 dose was determined to be 3.4 mg/kg based on activity and safety data. In part II, the most common adverse events at any grade were blurred vision, dry eye, thrombocytopenia, anemia, increased aspartate aminotransferase, and cough. The most frequent grade 3–4 adverse events were thrombocytopenia (35%), anemia (17%), pneumonia (6%), and infusion-related reactions (6%).
Belantamab mafodotin 由人源化阿福糖基化的 Fc 工程靶向 B 细胞成熟抗原 (BCMA) 的 IgG 组成,通过不可切割的马来酰亚胺卡缬酰 (mc) 接头与 MMAF 有效载荷共价连接。首次人体1期DREAMM-1临床试验探讨了belantamab mafodotin作为复发或难治性多发性骨髓瘤(RRMM)的单一疗法[117]。本研究包括两部分:(I) 剂量递增阶段,评估 belantamab mafodotin 的安全性和耐受性并确定推荐的 2 期剂量;(II) 剂量扩展阶段,评估推荐的 2 期剂量的安全性和耐受性、药代动力学和临床活性。在第一部分中,38 名患者每三周接受 0.03 至 4.6 mg/kg belantamab mafodotin。治疗耐受性良好,未确定最大耐受剂量。根据活性和安全性数据,推荐的 2 期剂量确定为 3.4 mg/kg。在第二部分中,任何级别最常见的不良事件是视力模糊、干眼症、血小板减少症、贫血、天冬氨酸转氨酶升高和咳嗽。最常见的 3-4 级不良事件是血小板减少症 (35%)、贫血 (17%)、肺炎 (6%) 和输液相关反应 (6%)。
Belantamab mafodotin 由人源化阿福糖基化的 Fc 工程靶向 B 细胞成熟抗原 (BCMA) 的 IgG 组成,通过不可切割的马来酰亚胺卡缬酰 (mc) 接头与 MMAF 有效载荷共价连接。首次人体1期DREAMM-1临床试验探讨了belantamab mafodotin作为复发或难治性多发性骨髓瘤(RRMM)的单一疗法[117]。本研究包括两部分:(I) 剂量递增阶段,评估 belantamab mafodotin 的安全性和耐受性并确定推荐的 2 期剂量;(II) 剂量扩展阶段,评估推荐的 2 期剂量的安全性和耐受性、药代动力学和临床活性。在第一部分中,38 名患者每三周接受 0.03 至 4.6 mg/kg belantamab mafodotin。治疗耐受性良好,未确定最大耐受剂量。根据活性和安全性数据,推荐的 2 期剂量确定为 3.4 mg/kg。在第二部分中,任何级别最常见的不良事件是视力模糊、干眼症、血小板减少症、贫血、天冬氨酸转氨酶升高和咳嗽。最常见的 3-4 级不良事件是血小板减少症 (35%)、贫血 (17%)、肺炎 (6%) 和输液相关反应 (6%)。
The phase 2 DREAMM-2 study assessed the safety and efficacy of belantamab mafodotin in patients with relapsed or refractory multiple myeloma via two dosage cohorts: 2.5 mg/kg (97 patients) and 3.4 mg/kg (99 patients) once every three weeks [118]. The most common adverse events (≥20%) were keratopathy occurring in 72% of patients in the 2.5 mg/kg cohort and 77% in the 3.4 mg/kg cohort, thrombocytopenia (35% and 58%), anemia (24% and 37%), nausea (24% and 32%), pyrexia (22% and 25%), blurred vision (22% and 30%), and increased aspartate aminotransferase (20% and 24%). The most common grade 3–4 adverse events were keratopathy (27% in the 2.5 mg/kg cohort and 21% in the 3.4 mg/kg cohort), thrombocytopenia (20% and 33%), and anemia (20% and 25%). Dose delays and dose reduction due to treatment-related adverse events occurred mainly due to keratopathy (47% in the 2.5 mg/kg cohort and 53% in the 3.4 mg/kg cohort) and (25% vs. 30%), respectively. Treatment discontinuation due to ocular toxicities occurred in 1% vs. 3% of patients. Due to the prevalence of ocular adverse events, topical steroid prophylaxis was utilized before the treatment of belantamab mafodotin to prevent ocular toxicities; however, it was not proven beneficial.
2期DREAMM-2研究通过2.5mg/kg(97例患者)和3.4mg/kg(99例患者)两个剂量队列,评估了belantamab mafodotin在复发或难治性多发性骨髓瘤患者中的安全性和有效性,每3周一次[118]。最常见的不良事件(≥20%)是2.5 mg/kg组72%的患者和3.4 mg/kg组77%的患者发生角膜病变、血小板减少症(35%和58%)、贫血(24%和37%)、恶心(24%和32%)、发热(22%和25%)、视力模糊(22%和30%)以及天冬氨酸转氨酶升高(20%和24%)。最常见的 3-4 级不良事件是角膜病变(2.5 mg/kg 队列为 27%,3.4 mg/kg 队列为 21%)、血小板减少症(20% 和 33%)和贫血(20% 和 25%)。治疗相关不良事件导致的剂量延迟和剂量减少主要由角膜病变引起(2.5 mg/kg组为47%,3.4 mg/kg组为53%)和(25%对30%)。因眼部毒性而停止治疗的患者发生率为 1% vs. 3%。由于眼部不良事件的普遍存在,在治疗belantamab mafodotin之前使用局部类固醇预防来预防眼毒性;然而,它并没有被证明是有益的。
2期DREAMM-2研究通过2.5mg/kg(97例患者)和3.4mg/kg(99例患者)两个剂量队列,评估了belantamab mafodotin在复发或难治性多发性骨髓瘤患者中的安全性和有效性,每3周一次[118]。最常见的不良事件(≥20%)是2.5 mg/kg组72%的患者和3.4 mg/kg组77%的患者发生角膜病变、血小板减少症(35%和58%)、贫血(24%和37%)、恶心(24%和32%)、发热(22%和25%)、视力模糊(22%和30%)以及天冬氨酸转氨酶升高(20%和24%)。最常见的 3-4 级不良事件是角膜病变(2.5 mg/kg 队列为 27%,3.4 mg/kg 队列为 21%)、血小板减少症(20% 和 33%)和贫血(20% 和 25%)。治疗相关不良事件导致的剂量延迟和剂量减少主要由角膜病变引起(2.5 mg/kg组为47%,3.4 mg/kg组为53%)和(25%对30%)。因眼部毒性而停止治疗的患者发生率为 1% vs. 3%。由于眼部不良事件的普遍存在,在治疗belantamab mafodotin之前使用局部类固醇预防来预防眼毒性;然而,它并没有被证明是有益的。
Ocular toxicity was included in the box warning in the package insert of the commercial belantamab mafodotin [119]. Ocular adverse reactions occurred in 77% of the 218 patients in the pooled safety population, including keratopathy (76%), changes in visual acuity (55%), blurred vision (27%), and dry eye (19%). Most keratopathy events developed within the first two treatment cycles (cumulative incidence of 65% by cycle 2).
眼毒性包含在商业belantamab mafodotin包装说明书的包装警告中[119]。在汇总安全人群中的 218 例患者中,77% 发生了眼部不良反应,包括角膜病变 (76%)、视力改变 (55%)、视力模糊 (27%) 和干眼症 (19%)。大多数角膜病变事件在前两个治疗周期内发生(第 2 周期的累积发病率为 65%)。
眼毒性包含在商业belantamab mafodotin包装说明书的包装警告中[119]。在汇总安全人群中的 218 例患者中,77% 发生了眼部不良反应,包括角膜病变 (76%)、视力改变 (55%)、视力模糊 (27%) 和干眼症 (19%)。大多数角膜病变事件在前两个治疗周期内发生(第 2 周期的累积发病率为 65%)。
Based on the results from the DREAMM-2 study, belantamab mafodotin was granted an accelerated approval by the FDA as monotherapy for patients with relapsed or refractory multiple myeloma. However, the ADC was voluntarily withdrawn from the US market in November 2022 due to its failure to meet the objective outcome of the phase 3 DREAMM-3 study [120]. Currently, belantamab mafodotin is under evaluation as combination therapy with chemotherapeutic agents for RRMM treatment in DREAMM-7 and DREAMM-8 confirmatory clinical trials.
根据 DREAMM-2 研究结果,belantamab mafodotin 被 FDA 加速批准用于复发或难治性多发性骨髓瘤患者的单一疗法。然而,由于未能达到3期DREAMM-3研究的客观结果,该ADC于2022年11月自愿退出美国市场[120]。目前,belantamab mafodotin 正在 DREAMM-7 和 DREAMM-8 验证性临床试验中作为化疗药物联合治疗 RRMM 治疗的评估。
根据 DREAMM-2 研究结果,belantamab mafodotin 被 FDA 加速批准用于复发或难治性多发性骨髓瘤患者的单一疗法。然而,由于未能达到3期DREAMM-3研究的客观结果,该ADC于2022年11月自愿退出美国市场[120]。目前,belantamab mafodotin 正在 DREAMM-7 和 DREAMM-8 验证性临床试验中作为化疗药物联合治疗 RRMM 治疗的评估。
3.1.3. ADCs with Maytansinoid Payloads
3.1.3. 具有Maytansinoid有效载荷的ADC
Trastuzumab Emtansine (Kadcyla)
曲妥珠单抗 Emtansine (Kadcyla)
Trastuzumab emtansine comprises the humanized anti-human epidermal growth factor receptor 2 (HER-2) trastuzumab, a DM1 cytotoxic payload, and an SMCC non-cleavable linker [121]. The initial phase 1 dose-escalation study assessed the safety, tolerability, and pharmacokinetics of trastuzumab emtansine in patients with locally advanced or metastatic HER2-positive breast cancer [122,123]. Two administration schedules, once every three weeks and once weekly, were evaluated. In the cohort that received treatment once every three weeks, 24 patients were treated with 0.3 to 4.8 mg/kg of trastuzumab emtansine, and the dose of 3.6 mg/kg was determined to be the maximum tolerated dose. The most common treatment-related adverse events of any grade that occurred in these patients were thrombocytopenia (54%), elevated transaminases (42%), fatigue (38%), anemia (29%), and nausea (25%). In the cohort that received trastuzumab emtansine doses of 1.2 to 2.9 mg/kg once weekly (n = 28), the maximum tolerated dose was 2.4 mg/kg. Grade ≥3 adverse events occurred in 68% of patients, of which anemia (14%), thrombocytopenia (11%), pneumonia (11%), and increased AST (11%) were the most common.
曲妥珠单抗emtansine包括人源化抗人表皮生长因子受体2(HER-2)曲妥珠单抗、DM1细胞毒性有效载荷和SMCC不可切割接头[121]。最初的1期剂量递增研究评估了曲妥珠单抗在局部晚期或转移性HER2阳性乳腺癌患者中的安全性、耐受性和药代动力学[122,123]。评估了两个给药时间表,每三周一次,每周一次。在每三周接受一次治疗的队列中,24 例患者接受了 0.3 至 4.8 mg/kg 曲妥珠单抗 emtansine 治疗,3.6 mg/kg 的剂量被确定为最大耐受剂量。这些患者中最常见的任何级别的治疗相关不良事件是血小板减少症 (54%)、转氨酶升高 (42%)、疲劳 (38%)、贫血 (29%) 和恶心 (25%)。在每周一次接受曲妥珠单抗 emtansine 剂量为 1.2 至 2.9 mg/kg 的队列中 (n = 28),最大耐受剂量为 2.4 mg/kg。68%的患者发生≥3级不良事件,其中贫血(14%)、血小板减少症(11%)、肺炎(11%)和AST升高(11%)最常见。
曲妥珠单抗emtansine包括人源化抗人表皮生长因子受体2(HER-2)曲妥珠单抗、DM1细胞毒性有效载荷和SMCC不可切割接头[121]。最初的1期剂量递增研究评估了曲妥珠单抗在局部晚期或转移性HER2阳性乳腺癌患者中的安全性、耐受性和药代动力学[122,123]。评估了两个给药时间表,每三周一次,每周一次。在每三周接受一次治疗的队列中,24 例患者接受了 0.3 至 4.8 mg/kg 曲妥珠单抗 emtansine 治疗,3.6 mg/kg 的剂量被确定为最大耐受剂量。这些患者中最常见的任何级别的治疗相关不良事件是血小板减少症 (54%)、转氨酶升高 (42%)、疲劳 (38%)、贫血 (29%) 和恶心 (25%)。在每周一次接受曲妥珠单抗 emtansine 剂量为 1.2 至 2.9 mg/kg 的队列中 (n = 28),最大耐受剂量为 2.4 mg/kg。68%的患者发生≥3级不良事件,其中贫血(14%)、血小板减少症(11%)、肺炎(11%)和AST升高(11%)最常见。
In the phase III randomized open-label clinical study EMILIA, 991 patients with unresectable or metastatic breast cancer, who had previously been treated with trastuzumab and a taxane, were given either trastuzumab emtansine (3.6 mg/kg every three weeks) or with lapatinib plus capecitabine [124]. The most common treatment-related adverse events of any grade in the trastuzumab emtansine group were nausea (39.2%), fatigue (35.1%), and thrombocytopenia (28.0%). Grade ≥ 3 adverse events occurred in 43.1% of patients, with thrombocytopenia (14%), increased aspartate aminotransferase levels (5%), and anemia (4%) being the most frequent. Dose delays occurred in 23.7% of patients, commonly due to neutropenia, thrombocytopenia, leukopenia, fatigue, increased transaminases, and pyrexia. Dose reductions were reported in 16.3% of patients, primarily due to thrombocytopenia, increased transaminases, and peripheral neuropathy. Treatment discontinuation occurred in 6.5% of patients, with thrombocytopenia and elevated transaminases being the most common causes.
在III.期随机开放标签临床研究EMILIA中,991例既往接受过曲妥珠单抗和紫杉烷治疗的不可切除或转移性乳腺癌患者,接受了曲妥珠单抗emtansine(3.6mg/kg,每3周一次)或拉帕替尼联合卡培他滨[124]。曲妥珠单抗emtansine组任何级别最常见的治疗相关不良事件是恶心(39.2%)、疲劳(35.1%)和血小板减少症(28.0%)。43.1% 的患者发生 ≥ 3 级不良事件,最常见的是血小板减少症 (14%)、天冬氨酸转氨酶水平升高 (5%) 和贫血 (4%)。23.7% 的患者出现剂量延迟,通常是由于中性粒细胞减少、血小板减少、白细胞减少、疲劳、转氨酶升高和发热。据报道,16.3% 的患者剂量减少,主要是由于血小板减少、转氨酶升高和周围神经病变。6.5%的患者停止治疗,血小板减少症和转氨酶升高是最常见的原因。
在III.期随机开放标签临床研究EMILIA中,991例既往接受过曲妥珠单抗和紫杉烷治疗的不可切除或转移性乳腺癌患者,接受了曲妥珠单抗emtansine(3.6mg/kg,每3周一次)或拉帕替尼联合卡培他滨[124]。曲妥珠单抗emtansine组任何级别最常见的治疗相关不良事件是恶心(39.2%)、疲劳(35.1%)和血小板减少症(28.0%)。43.1% 的患者发生 ≥ 3 级不良事件,最常见的是血小板减少症 (14%)、天冬氨酸转氨酶水平升高 (5%) 和贫血 (4%)。23.7% 的患者出现剂量延迟,通常是由于中性粒细胞减少、血小板减少、白细胞减少、疲劳、转氨酶升高和发热。据报道,16.3% 的患者剂量减少,主要是由于血小板减少、转氨酶升高和周围神经病变。6.5%的患者停止治疗,血小板减少症和转氨酶升高是最常见的原因。
Hepatotoxicity is included in one of the black box warnings for trastuzumab emtansine treatment, presenting as asymptomatic transient increases of serum transaminases [66]. Liver failure and fatal cases of severe treatment-related liver injury have been reported. The hepatotoxicity associated with trastuzumab emtansine treatment might be due to the HER2-dependent and independent pathways. HER2 is expressed in normal human hepatocytes, and HER-2 mediated internalization of trastuzumab emtansine causes cell-cycle arrest and apoptosis in human hepatocytes in vitro [125,126]. In addition, DM1/DM4 payloads have been shown to mediate the internalization of maytansinoid-conjugated ADCs via binding to cytoskeleton-associated protein 5 (CKAP5) [127,128]. Receptor-mediated uptake of the ADC by FcγRs and mannose receptors may also contribute to liver toxicity [32]. Besides hepatotoxicity, cardiac toxicity and embryo-fetal toxicity are also included in the black box warning. Cardiac toxicity manifests as a decrease in left ventricular ejection fraction (LVEF) to less than 40%, which is related to the trastuzumab component [129]. Moreover, several complications, including oligohydramnios and fetal/neonatal death, have been associated with trastuzumab exposure during the second or third trimester of pregnancy [11,130,131].
曲妥珠单抗治疗的黑匣子警告之一包括肝毒性,表现为血清转氨酶无症状短暂性升高[66]。肝衰竭和严重治疗相关肝损伤的致命病例已有报道。与曲妥珠单抗emtansine治疗相关的肝毒性可能是由于HER2依赖性和独立途径。HER2在正常人肝细胞中表达,HER-2介导的曲妥珠单抗emtansine内化在体外引起人肝细胞的细胞周期停滞和凋亡[125,126]。此外,DM1/DM4有效载荷已被证明通过与细胞骨架相关蛋白5(CKAP5)结合来介导美登素偶联ADC的内化[127,128]。受体介导的FcγR和甘露糖受体对ADC的摄取也可能导致肝毒性[32]。除肝毒性外,心脏毒性和胚胎-胎儿毒性也包含在黑匣子警告中。心脏毒性表现为左心室射血分数(left ventricular ejection fraction, LVEF)降低至40%以下,这与曲妥珠单抗组分有关[129]。此外,一些并发症,包括羊水过少和胎儿/新生儿死亡,与妊娠中期或晚期曲妥珠单抗暴露有关[11,130,131]。
曲妥珠单抗治疗的黑匣子警告之一包括肝毒性,表现为血清转氨酶无症状短暂性升高[66]。肝衰竭和严重治疗相关肝损伤的致命病例已有报道。与曲妥珠单抗emtansine治疗相关的肝毒性可能是由于HER2依赖性和独立途径。HER2在正常人肝细胞中表达,HER-2介导的曲妥珠单抗emtansine内化在体外引起人肝细胞的细胞周期停滞和凋亡[125,126]。此外,DM1/DM4有效载荷已被证明通过与细胞骨架相关蛋白5(CKAP5)结合来介导美登素偶联ADC的内化[127,128]。受体介导的FcγR和甘露糖受体对ADC的摄取也可能导致肝毒性[32]。除肝毒性外,心脏毒性和胚胎-胎儿毒性也包含在黑匣子警告中。心脏毒性表现为左心室射血分数(left ventricular ejection fraction, LVEF)降低至40%以下,这与曲妥珠单抗组分有关[129]。此外,一些并发症,包括羊水过少和胎儿/新生儿死亡,与妊娠中期或晚期曲妥珠单抗暴露有关[11,130,131]。
Mirvetuximab Soravtansine (Elahere)
Mirvetuximab soravtansine is an ADC composed of a humanized antibody targeting the folate receptor alpha (FRα), a disulfide cleavable linker, and a DM4 cytotoxic payload [132]. In the phase 1 dose-escalation, 44 patients with advanced solid tumors were treated with mirvertuximab soravtansine at doses between 0.15 mg/kg and 7 mg/kg based on total body weight (TBW) once every three weeks [133]. At the 7 mg/kg dose level, 1 of 5 patients had a DLT of punctate keratitis. At the 5 mg/kg dose level in 11 patients, grade 3 hypophosphatemia and additional ocular toxicities were observed. Subsequently, dosing was modified based on adjusted ideal body weight (AIBW) rather than TBW, and no DLT was observed in patients given up to 6 mg/kg AIBW, which was determined to be the recommended phase 2 dose.
In the phase 3 open-label randomized clinical trial FORWARD I, 366 patients with FRα-positive platinum-resistant ovarian cancer were given either mirvetuximab soravtansine (n = 243) at a 6 mg/kg AIBW dose or chemotherapy (n = 109) [134]. In the mirvetuximab soravtansine group, the most common treatment-related adverse events were nausea (45.7%), blurred vision (42.0%), keratopathy (32.5%), diarrhea (31.3%), fatigue (28.8%), peripheral neuropathy (26.7%), dry eye (25.9%), and decreased visual acuity (19.3%). Incidents of grade ≥ 3 treatment-related adverse events were low, with blurred vision (2.5%), peripheral neuropathy (2.5%), and diarrhea (2.1%) being the most common. Dose delays/reductions occurred in 34.3% of patients, with ocular toxicity being the most common cause (19.8%). Dose discontinuations occurred in 4.5% of patients.
3.1.4. ADCs with Camptothecin Payloads
Trastuzumab Deruxtecan (Enhertu)
Trastuzumab deruxtecan is a humanized anti-HER2 antibody linked to deruxtecan, a camptothecin derivative, via a stable linker [135]. In the first phase 1 dose-escalation study, 22 patients with HER-2 positive advanced or metastatic breast cancer, gastric cancer, or other HER-2 expressing solid tumors were treated with 0.8 mg/kg to 8.0 mg/kg of trastuzumab deruxtecan once every three weeks [136]. No dose-limiting toxicity was observed, and MTD was not reached. Target drug exposure was achieved at the dose of 6.4 mg/kg, which was selected as the recommended phase 2 dose.
The pivotal single-arm phase 2 DESTINY-Breast01 clinical trial consisted of two parts [65]. In the first part, advanced/metastatic breast cancer patients previously treated with more than 2 anti-HER2 therapies were randomized to receive trastuzumab deruxtecan at doses of 5.4 mg/kg (n = 50), 6.4 mg/kg (n = 48), or 7.4 mg/kg (n = 21) once every 3 weeks. In the second part, 134 patients were treated with 5.4 mg/kg doses based on the obtained efficacy and toxicity data from part 1. Among the 184 patients treated with trastuzumab deruxtecan at 5.4 mg/kg, the most common adverse events of all grades (≥20%) were nausea (77.5%), fatigue (49.8%), alopecia (49.8%), vomiting (44.3%), neutropenia (40.3%), constipation (37.5%), anemia (33.6%), decreased appetite (33.2%), diarrhea (29.2%), leukopenia (26.9%), and thrombocytopenia (24.9%). Grade ≥3 adverse events occurred in 57.1% of the patients, with neutropenia (20.7%), anemia (8.7%), nausea (7.6%), leukopenia (6.5%), lymphopenia (6.5%), and fatigue (6.0%) being the most prevalent. Dose interruption, dose reduction, and treatment discontinuation due to adverse events occurred in 35.3%, 23.4%, and 15.2% of patients, respectively, with pneumonitis (in 11 patients) and interstitial lung disease (in 5 patients) being the most common reasons. Interstitial lung disease (ILD) and pneumonitis are included in the black box warning for patients treated with trastuzumab deruxtecan [137]. Treatment-related interstitial lung disease and fatal outcomes occurred in 9% and 2.6% of patients treated with trastuzumab deruxtecan, respectively. Similar to other HER-2 targeting ADCs, patients treated with trastuzumab deruxtecan also have increased risks of embryo-fetal toxicity and left ventricular dysfunction.
关键的单臂2期DESTINY-Breast01临床试验由两部分组成[65]。在第一部分中,先前接受过 2 种以上抗 HER2 疗法治疗的晚期/转移性乳腺癌患者被随机分配接受剂量为 5.4 mg/kg (n = 50)、6.4 mg/kg (n = 48) 或 7.4 mg/kg (n = 21) 的曲妥珠单抗 deruxtecan,每 3 周一次。在第二部分中,根据从第一部分获得的疗效和毒性数据,对 134 名患者进行了 5.4 mg/kg 剂量的治疗。在5.4 mg/kg曲妥珠单抗治疗的184例患者中,所有级别最常见的不良事件(≥20%)为恶心(77.5%)、疲劳(49.8%)、脱发(49.8%)、呕吐(44.3%)、中性粒细胞减少(40.3%)、便秘(37.5%)、贫血(33.6%)、食欲下降(33.2%)、腹泻(29.2%)、白细胞减少(26.9%)和血小板减少(24.9%)。57.1%的患者发生≥3级不良事件,其中中性粒细胞减少(20.7%)、贫血(8.7%)、恶心(7.6%)、白细胞减少(6.5%)、淋巴细胞减少(6.5%)和疲劳(6.0%)最为普遍。35.3%、23.4%和15.2%的患者因不良事件导致剂量中断、剂量减少和停止治疗,其中肺炎(11例)和间质性肺病(5例)是最常见的原因。间质性肺病(interstitial lung disease, ILD)和肺炎被纳入曲妥珠单抗deruxtecan治疗患者的黑匣子警告中[ 137]。在接受曲妥珠单抗deruxtecan治疗的患者中,分别有9%和2.6%的患者发生治疗相关的间质性肺病和致死性结局。与其他靶向 HER-2 的 ADC 类似,接受曲妥珠单抗 deruxtecan 治疗的患者发生胚胎-胎儿毒性和左心室功能障碍的风险也会增加。
关键的单臂2期DESTINY-Breast01临床试验由两部分组成[65]。在第一部分中,先前接受过 2 种以上抗 HER2 疗法治疗的晚期/转移性乳腺癌患者被随机分配接受剂量为 5.4 mg/kg (n = 50)、6.4 mg/kg (n = 48) 或 7.4 mg/kg (n = 21) 的曲妥珠单抗 deruxtecan,每 3 周一次。在第二部分中,根据从第一部分获得的疗效和毒性数据,对 134 名患者进行了 5.4 mg/kg 剂量的治疗。在5.4 mg/kg曲妥珠单抗治疗的184例患者中,所有级别最常见的不良事件(≥20%)为恶心(77.5%)、疲劳(49.8%)、脱发(49.8%)、呕吐(44.3%)、中性粒细胞减少(40.3%)、便秘(37.5%)、贫血(33.6%)、食欲下降(33.2%)、腹泻(29.2%)、白细胞减少(26.9%)和血小板减少(24.9%)。57.1%的患者发生≥3级不良事件,其中中性粒细胞减少(20.7%)、贫血(8.7%)、恶心(7.6%)、白细胞减少(6.5%)、淋巴细胞减少(6.5%)和疲劳(6.0%)最为普遍。35.3%、23.4%和15.2%的患者因不良事件导致剂量中断、剂量减少和停止治疗,其中肺炎(11例)和间质性肺病(5例)是最常见的原因。间质性肺病(interstitial lung disease, ILD)和肺炎被纳入曲妥珠单抗deruxtecan治疗患者的黑匣子警告中[ 137]。在接受曲妥珠单抗deruxtecan治疗的患者中,分别有9%和2.6%的患者发生治疗相关的间质性肺病和致死性结局。与其他靶向 HER-2 的 ADC 类似,接受曲妥珠单抗 deruxtecan 治疗的患者发生胚胎-胎儿毒性和左心室功能障碍的风险也会增加。
Sacituzumab Govitecan (Trodelvy)
Sacituzumab Govitecan(Trodelvy)
Sacituzumab govitecan is a humanized anti-TROP-2 IgG linked to the active metabolite of irinotecan (SN-38) via a pH-sensitive linker [138]. In phase 1 of the first-in-human dose-escalation dose-expansion phase 1/2 study, 25 patients with diverse metastatic solid tumors were treated with 8 mg/kg to 18 mg/kg of sacituzumab govitecan on days 1 and 8 of 21-day cycles [139]. The MTD was determined to be 12 mg/kg for the first cycle, with neutropenia being the dose-limiting toxicity. However, this dose level is too toxic for subsequent cycles; therefore, the 8 mg/kg and 10 mg/kg doses were selected for the phase 2 study. In phase 2 of this study, patients with diverse metastatic epithelial cancers who had multiple prior therapies received sacituzumab govitecan at either an 8 mg/kg (n = 81) or a 10 mg/kg (n = 97) dose [140]. The most common adverse events of all grades (≥25%) reported in the 8-mg/kg and 10-mg/kg cohorts were nausea (59% vs. 63%), diarrhea (53% vs. 62%), neutropenia (42% vs. 58%), fatigue (61% vs. 52%), vomiting (36% vs. 43%), anemia (38% vs. 42%), alopecia (46% vs. 37%), and constipation (33% vs. 37%), respectively. The most common adverse events of grades ≥3 (≥10%) reported in the 8-mg/kg and 10-mg/kg cohorts were neutropenia (30% vs. 36%), anemia (13% vs. 12%), diarrhea (4% vs. 10%), and leukopenia (6% vs. 12%). Dose reductions occurred in 19% and 28% of the patients in the 8-mg/kg and 10-mg/kg cohorts, respectively. Neutropenia was the most common adverse event leading to dose delays or reductions. Significantly more patients in the 10-mg/kg cohort experienced grade ≥3 neutropenia after the first dose than in the 8-mg/kg cohort (47% vs. 21%).
Sacituzumab govitecan是一种人源化抗TROP-2 IgG,通过pH敏感接头与伊立替康(SN-38)的活性代谢物相连[138]。在首次人体剂量递增剂量扩展1/2期研究的1期研究中,25例不同转移性实体瘤患者在21日周期的第1天和第8天接受8mg/kg至18mg/kg的sacituzumab govitecan治疗[139]。第一个周期的 MTD 确定为 12 mg/kg,中性粒细胞减少是剂量限制性毒性。然而,这个剂量水平对于后续周期来说毒性太大;因此,8 mg/kg 和 10 mg/kg 剂量被选为 2 期研究。在这项研究的第2阶段,既往接受过多种治疗的多发转移性上皮癌患者接受了8mg/kg(n=81)或10mg/kg(n=97)剂量的sacituzumab govitecan治疗[140]。在8 mg/kg和10 mg/kg队列中报告的所有等级(≥25%)最常见的不良事件分别是恶心(59% vs. 63%)、腹泻(53% vs. 62%)、中性粒细胞减少(42% vs. 58%)、疲劳(61% vs. 52%)、呕吐(36% vs. 43%)、贫血(38% vs. 42%)、脱发(46% vs. 37%)和便秘(33% vs. 37%)。在 8 mg/kg 和 10 mg/kg 队列中报告的 ≥3 级 (≥10%) 最常见的不良事件是中性粒细胞减少(30% vs. 36%)、贫血(13% vs. 12%)、腹泻(4% vs. 10%)和白细胞减少(6% vs. 12%)。在 8 mg/kg 和 10 mg/kg 队列中,分别有 19% 和 28% 的患者发生剂量减少。中性粒细胞减少是导致剂量延迟或减少的最常见不良事件。10 mg/kg 队列中出现 ≥3 级中性粒细胞减少的患者在首次给药后明显多于 8 mg/kg 队列(47% vs. 21%)。
Sacituzumab govitecan是一种人源化抗TROP-2 IgG,通过pH敏感接头与伊立替康(SN-38)的活性代谢物相连[138]。在首次人体剂量递增剂量扩展1/2期研究的1期研究中,25例不同转移性实体瘤患者在21日周期的第1天和第8天接受8mg/kg至18mg/kg的sacituzumab govitecan治疗[139]。第一个周期的 MTD 确定为 12 mg/kg,中性粒细胞减少是剂量限制性毒性。然而,这个剂量水平对于后续周期来说毒性太大;因此,8 mg/kg 和 10 mg/kg 剂量被选为 2 期研究。在这项研究的第2阶段,既往接受过多种治疗的多发转移性上皮癌患者接受了8mg/kg(n=81)或10mg/kg(n=97)剂量的sacituzumab govitecan治疗[140]。在8 mg/kg和10 mg/kg队列中报告的所有等级(≥25%)最常见的不良事件分别是恶心(59% vs. 63%)、腹泻(53% vs. 62%)、中性粒细胞减少(42% vs. 58%)、疲劳(61% vs. 52%)、呕吐(36% vs. 43%)、贫血(38% vs. 42%)、脱发(46% vs. 37%)和便秘(33% vs. 37%)。在 8 mg/kg 和 10 mg/kg 队列中报告的 ≥3 级 (≥10%) 最常见的不良事件是中性粒细胞减少(30% vs. 36%)、贫血(13% vs. 12%)、腹泻(4% vs. 10%)和白细胞减少(6% vs. 12%)。在 8 mg/kg 和 10 mg/kg 队列中,分别有 19% 和 28% 的患者发生剂量减少。中性粒细胞减少是导致剂量延迟或减少的最常见不良事件。10 mg/kg 队列中出现 ≥3 级中性粒细胞减少的患者在首次给药后明显多于 8 mg/kg 队列(47% vs. 21%)。
Black box warnings were added to the sacituzumab govitecan label for severe or life-threatening neutropenia and severe diarrhea [141]. These adverse events are likely mediated by released (“free”) SN-38, as the same toxicities are associated with the SN-38 prodrug ironotecan [142]. Of all patients treated with sacituzumab govitecan, neutropenia of all grades and grade ≥ 3 occurred in 61% and 47%, respectively. Febrile neutropenia occurred in 7% of patients. Diarrhea of all grades and grade ≥ 3 occurred in 65% and 12% of all patients treated sacituzumab govitecan, respectively. Neutropenic colitis occurred in 0.5% of patients.
sacituzumab govitecan标签上增加了黑匣子警告,用于严重或危及生命的中性粒细胞减少症和严重腹泻[141]。这些不良事件可能是由释放(“游离”)SN-38介导的,因为SN-38前药伊洛替康具有相同的毒性[142]。在接受sacituzumab govitecan治疗的所有患者中,所有级别和≥3级的中性粒细胞减少症分别发生率为61%和47%。发热性中性粒细胞减少症发生在 7% 的患者中。所有级别和≥ 3级腹泻分别发生于所有接受sacituzumab govitecan治疗的患者中,有65%和12%发生腹泻。中性粒细胞减少性结肠炎发生率为0.5%。
sacituzumab govitecan标签上增加了黑匣子警告,用于严重或危及生命的中性粒细胞减少症和严重腹泻[141]。这些不良事件可能是由释放(“游离”)SN-38介导的,因为SN-38前药伊洛替康具有相同的毒性[142]。在接受sacituzumab govitecan治疗的所有患者中,所有级别和≥3级的中性粒细胞减少症分别发生率为61%和47%。发热性中性粒细胞减少症发生在 7% 的患者中。所有级别和≥ 3级腹泻分别发生于所有接受sacituzumab govitecan治疗的患者中,有65%和12%发生腹泻。中性粒细胞减少性结肠炎发生率为0.5%。
3.1.5. ADCs with Pyrrolobenzodiazepine Payloads
3.1.5. 具有吡咯苯并二氮卓有效载荷的ADC
Loncastuximab Tesirine (Zynlonta)
Loncastuximab Tesirine(Zynlonta)
Loncastuximab tesirine comprises a humanized anti-CD19 IgG, a valine-alanine protease cleavable linker, and a PBD DNA-alkylating cytotoxic payload [143,144]. In the dose-escalation dose-expansion phase 1 study, 183 patients with relapsed/refractory B-cell non-Hodgkin lymphoma received 15 µg/kg to 200 µg/kg of loncastuximab tesirine once every three weeks [145]. Dose-limiting toxicities included grade 4 thrombocytopenia and grade 3 febrile neutropenia, and the maximum tolerated dose was not reached; however, cumulative toxicity was observed at 200 µg/kg. The 150 µg/kg dose was selected as the recommended phase 2 dose, as it elicited encouraging responses and a lower frequency of adverse events than the 200 µg/kg dose. In addition, due to a moderate accumulation of loncastuximab tesirine, late-developing and difficult-to-manage toxicities frequently led to dose delays and dose reductions; therefore, the recommended phase 2 dose includes a planned dose reduction to 75 µg/kg after two cycles.
Loncastuximab tesirine 包括人源化抗 CD19 IgG、缬氨酸-丙氨酸蛋白酶可裂解接头和 PBD DNA 烷化细胞毒性有效载荷 [ 143, 144]。在剂量递增剂量扩展1期研究中,183例复发/难治性B细胞非霍奇金淋巴瘤患者每3周一次接受15μg/kg-200μg/kg的loncastuximab tesirine治疗[145]。剂量限制性毒性包括 4 级血小板减少症和 3 级发热性中性粒细胞减少症,未达到最大耐受剂量;然而,在200 μg/kg时观察到累积毒性。150 μg/kg剂量被选为推荐的2期剂量,因为它引起了令人鼓舞的反应,并且与200 μg/kg剂量相比,不良事件的发生频率更低。此外,由于loncastuximab tesirine的适度积累,晚期发展和难以管理的毒性经常导致剂量延迟和剂量减少;因此,推荐的 2 期剂量包括两个周期后计划将剂量减少至 75 μg/kg。
Loncastuximab tesirine 包括人源化抗 CD19 IgG、缬氨酸-丙氨酸蛋白酶可裂解接头和 PBD DNA 烷化细胞毒性有效载荷 [ 143, 144]。在剂量递增剂量扩展1期研究中,183例复发/难治性B细胞非霍奇金淋巴瘤患者每3周一次接受15μg/kg-200μg/kg的loncastuximab tesirine治疗[145]。剂量限制性毒性包括 4 级血小板减少症和 3 级发热性中性粒细胞减少症,未达到最大耐受剂量;然而,在200 μg/kg时观察到累积毒性。150 μg/kg剂量被选为推荐的2期剂量,因为它引起了令人鼓舞的反应,并且与200 μg/kg剂量相比,不良事件的发生频率更低。此外,由于loncastuximab tesirine的适度积累,晚期发展和难以管理的毒性经常导致剂量延迟和剂量减少;因此,推荐的 2 期剂量包括两个周期后计划将剂量减少至 75 μg/kg。
In the pivotal phase 2 LOTIS-2 clinical trial, 145 patients with relapsed or refractory diffuse large B-cell lymphoma were treated with loncastuximab tesirine once every three weeks at 150 µg/kg for two cycles, and then at 75 μg/kg after that [146]. The most common treatment-related adverse events of all grades (≥25%) were neutropenia (40%), thrombocytopenia (33%), anemia (26%), fatigue (27%), and gamma-glutamyl transferase increase (40%). The most common grade ≥3 treatment-related adverse events (≥5%) were thrombocytopenia (18%), neutropenia (26%), anemia (10%), gamma-glutamyl transferase increase (16%), leukopenia (9%), lymphopenia (5%), and hypophosphatemia (6%). Serious adverse reactions occurred in 28% of patients, with febrile neutropenia, pneumonia, edema, pleural effusion, and sepsis being the most common (≥2%). Fatal adverse reactions occurred in 1% of patients due to infection. Dose delays occurred in 49% of patients, with gamma-glutamyl transferase increase, neutropenia, thrombocytopenia, and edema being the most common reasons (≥5%). Dose reductions occurred in 8% of patients, with the gamma-glutamyltransferase increase being the most common reason (≥4%). Treatment discontinuation occurred 19% of patients, due to gamma-glutamyltransferase increase, edema, and effusion (≥2%) [147].
在关键的2期LOTIS-2临床试验中,145例复发或难治性弥漫性大B细胞淋巴瘤患者每3周接受loncastuximab tesirine治疗,剂量为150μg/kg,持续两个周期,之后以75μg/kg治疗[146]。所有级别(≥25%)最常见的治疗相关不良事件是中性粒细胞减少(40%)、血小板减少(33%)、贫血(26%)、疲劳(27%)和γ-谷氨酰转移酶升高(40%)。最常见的 ≥3 级治疗相关不良事件 (≥5%) 为血小板减少症 (18%)、中性粒细胞减少症 (26%)、贫血 (10%)、γ-谷氨酰转移酶升高 (16%)、白细胞减少症 (9%)、淋巴细胞减少症 (5%) 和低磷血症 (6%)。28%的患者发生严重不良反应,其中发热性中性粒细胞减少、肺炎、水肿、胸腔积液和脓毒症最常见(≥2%)。1%的患者因感染而发生致命不良反应。49%的患者出现剂量延迟,γ-谷氨酰转移酶升高、中性粒细胞减少、血小板减少和水肿是最常见的原因(≥5%)。8% 的患者出现剂量减少,γ-谷氨酰转移酶升高是最常见的原因 (≥4%)。19%的患者因γ-谷氨酰转移酶升高、水肿和积液(≥2%)而停止治疗[147]。
在关键的2期LOTIS-2临床试验中,145例复发或难治性弥漫性大B细胞淋巴瘤患者每3周接受loncastuximab tesirine治疗,剂量为150μg/kg,持续两个周期,之后以75μg/kg治疗[146]。所有级别(≥25%)最常见的治疗相关不良事件是中性粒细胞减少(40%)、血小板减少(33%)、贫血(26%)、疲劳(27%)和γ-谷氨酰转移酶升高(40%)。最常见的 ≥3 级治疗相关不良事件 (≥5%) 为血小板减少症 (18%)、中性粒细胞减少症 (26%)、贫血 (10%)、γ-谷氨酰转移酶升高 (16%)、白细胞减少症 (9%)、淋巴细胞减少症 (5%) 和低磷血症 (6%)。28%的患者发生严重不良反应,其中发热性中性粒细胞减少、肺炎、水肿、胸腔积液和脓毒症最常见(≥2%)。1%的患者因感染而发生致命不良反应。49%的患者出现剂量延迟,γ-谷氨酰转移酶升高、中性粒细胞减少、血小板减少和水肿是最常见的原因(≥5%)。8% 的患者出现剂量减少,γ-谷氨酰转移酶升高是最常见的原因 (≥4%)。19%的患者因γ-谷氨酰转移酶升高、水肿和积液(≥2%)而停止治疗[147]。
3.2. Late-Stage ADCs 3.2. 后期ADC
3.2.1. Trastuzumab Duocarmazine
3.2.1. 曲妥珠单抗 Duocarmazine
Trastuzumab duocarmazine consists of the anti-HER-2 trastuzumab antibody linked to a duocamycin cytotoxic payload via a hydrophilic cleavable peptide linker [148]. In the dose-escalation study of phase 1 clinical trial, 39 patients with locally advanced or metastatic solid tumors, regardless of HER-2 expression, were given trastuzumab duocarmazine at 0.3 mg/kg to 2.4 mg/kg doses every three weeks [67]. One patient treated at 2.4 mg/kg died of pneumonitis, and no other DLT was observed. The 1.2 mg/kg dose once every three weeks was recommended for subsequent studies due to promising results in patients treated at this dose level, and the 1.5 mg/kg and 1.8 mg/kg doses did not improve the benefit-to-risk ratio for patients. In the dose-expansion study, 146 patients with breast cancer (n = 99), gastric cancer (n = 17), urothelial cancer (n = 16), and endometrial cancer (n = 14) were treated at the recommended phase 2 dose of 1.2 mg/kg every three weeks. The most common treatment-related adverse events were fatigue (33%), conjunctivitis (31%), dry eye (31%), lacrimation increase (20%), decreased appetite (19%), keratitis (19%), dry skin (18%), alopecia (18%), nausea (18%), stomatitis (16%), skin hyperpigmentation (16%), and neutropenia (16%). Ocular adverse events occurred in 71% of patients, including conjunctivitis, dry eye, keratitis, blurred vision, and corneal toxicity. Grade ≥ 3 treatment-related adverse events occurred in 35% of patients, with neutropenia (6%), fatigue (4%), and conjunctivitis (3%) being the most common. Dose delays/reductions occurred in 43% of patients, and treatment discontinuations occurred in 19% of patients, mostly due to ocular toxicity (10%). Other treatment-related toxicities that led to dose discontinuations were dyspnea, decreased LVEF, and decreased appetite.
曲妥珠单抗 duocarmazine 由抗 HER-2 曲妥珠单抗抗体组成,该抗体通过亲水性可裂解肽接头与多妥霉素细胞毒性有效载荷连接 [ 148]。在1期临床试验的剂量递增研究中,39例局部晚期或转移性实体瘤患者,无论HER-2表达如何,均以0.3mg/kg-2.4mg/kg的剂量给予曲妥珠单抗duocarmazine,每3周一次[67]。1例以2.4mg/kg治疗的患者死于肺炎,未观察到其他DLT。1.2 mg/kg 剂量被推荐用于后续研究,因为以该剂量水平治疗的患者有希望的结果,并且 1.5 mg/kg 和 1.8 mg/kg 剂量并没有改善患者的获益风险比。在剂量扩展研究中,146 名乳腺癌 (n = 99)、胃癌 (n = 17)、尿路上皮癌 (n = 16) 和子宫内膜癌 (n = 14) 患者以推荐的 2 期剂量每三周 1.2 mg/kg 进行治疗。最常见的治疗相关不良事件是疲劳(33%)、结膜炎(31%)、干眼症(31%)、流泪增加(20%)、食欲下降(19%)、角膜炎(19%)、皮肤干燥(18%)、脱发(18%)、恶心(18%)、口腔炎(16%)、皮肤色素沉着过度(16%)和中性粒细胞减少(16%)。71%的患者发生眼部不良事件,包括结膜炎、干眼症、角膜炎、视力模糊和角膜毒性。35% 的患者发生 ≥ 3 级治疗相关不良事件,其中中性粒细胞减少 (6%)、疲劳 (4%) 和结膜炎 (3%) 最常见。43%的患者出现剂量延迟/减少,19%的患者出现治疗中断,主要原因是眼部毒性(10%)。 其他导致停药的治疗相关毒性包括呼吸困难、LVEF 降低和食欲下降。
曲妥珠单抗 duocarmazine 由抗 HER-2 曲妥珠单抗抗体组成,该抗体通过亲水性可裂解肽接头与多妥霉素细胞毒性有效载荷连接 [ 148]。在1期临床试验的剂量递增研究中,39例局部晚期或转移性实体瘤患者,无论HER-2表达如何,均以0.3mg/kg-2.4mg/kg的剂量给予曲妥珠单抗duocarmazine,每3周一次[67]。1例以2.4mg/kg治疗的患者死于肺炎,未观察到其他DLT。1.2 mg/kg 剂量被推荐用于后续研究,因为以该剂量水平治疗的患者有希望的结果,并且 1.5 mg/kg 和 1.8 mg/kg 剂量并没有改善患者的获益风险比。在剂量扩展研究中,146 名乳腺癌 (n = 99)、胃癌 (n = 17)、尿路上皮癌 (n = 16) 和子宫内膜癌 (n = 14) 患者以推荐的 2 期剂量每三周 1.2 mg/kg 进行治疗。最常见的治疗相关不良事件是疲劳(33%)、结膜炎(31%)、干眼症(31%)、流泪增加(20%)、食欲下降(19%)、角膜炎(19%)、皮肤干燥(18%)、脱发(18%)、恶心(18%)、口腔炎(16%)、皮肤色素沉着过度(16%)和中性粒细胞减少(16%)。71%的患者发生眼部不良事件,包括结膜炎、干眼症、角膜炎、视力模糊和角膜毒性。35% 的患者发生 ≥ 3 级治疗相关不良事件,其中中性粒细胞减少 (6%)、疲劳 (4%) 和结膜炎 (3%) 最常见。43%的患者出现剂量延迟/减少,19%的患者出现治疗中断,主要原因是眼部毒性(10%)。 其他导致停药的治疗相关毒性包括呼吸困难、LVEF 降低和食欲下降。
In the phase 3 randomized TULIP trial, 437 patients with HER-2 positive locally advanced or metastatic breast cancer were treated with either trastuzumab duocarmazine (n = 291) at the dose of 1.2 mg/kg every three weeks or physician’s choice chemotherapy (n = 146) [149]. In the interim report, the most common reported adverse events for trastuzumab duocarmazine were conjunctivitis (38.2%), keratitis (38.2%), and fatigue (33.3%). Dose discontinuation occurred in 35.4% of patients treated with trastuzumab duocarmazine, mainly due to ocular toxicities (20.8%) and respiratory disorders (6.3%).
在3期随机TULIP试验中,437例HER-2阳性局部晚期或转移性乳腺癌患者接受曲妥珠单抗、多卡马嗪(n=291)治疗,剂量为1.2mg/kg/kg/3周,或医生选择化疗(n=146)[149]。在中期报告中,曲妥珠单抗多卡马嗪最常见的不良事件是结膜炎(38.2%)、角膜炎(38.2%)和疲劳(33.3%)。在接受曲妥珠单抗治疗的患者中,有35.4%的患者停药,主要是由于眼部毒性(20.8%)和呼吸系统疾病(6.3%)。
在3期随机TULIP试验中,437例HER-2阳性局部晚期或转移性乳腺癌患者接受曲妥珠单抗、多卡马嗪(n=291)治疗,剂量为1.2mg/kg/kg/3周,或医生选择化疗(n=146)[149]。在中期报告中,曲妥珠单抗多卡马嗪最常见的不良事件是结膜炎(38.2%)、角膜炎(38.2%)和疲劳(33.3%)。在接受曲妥珠单抗治疗的患者中,有35.4%的患者停药,主要是由于眼部毒性(20.8%)和呼吸系统疾病(6.3%)。
3.2.2. Disitamab Vedotin
Disitamab vedotin consists of a humanized anti-HER-2 mAb conjugated to an MMAE payload via a cleavable valine-citruline linker [150]. It was granted conditional approval in China in June 2021 and fast-track designations by the FDA in the US [151]. In the phase 1 dose-escalation study, 21 patients with HER2-overexpressed advanced solid cancers were treated with 0.1 mg/kg to 2.5 mg/kg of disitamab vedotin once every two weeks [152]. The most common treatment-related adverse events were leukopenia (61.1%), neutropenia (52.8%), fatigue (50.0%), numbness (44.4%), AST elevation (30.6%) and ALT elevation (27.8%). The maximum tolerated dose was not determined up to 2.5 mg/kg. From a pool of five phase 1–3 clinical trials in China, the most common treatment-related adverse events were leukopenia (55.4%), alopecia (54.6%), neutropenia (50.6%), increased aspartate aminotransferase (49.7%), fatigue (46.3%), increased alanine aminotransferase (42.9%), and hypoesthesia (40.9%) [151]. The most common grade ≥ 3 adverse events were neutropenia (16.9%), leukopenia (10.9%), and hypoesthesia (8.9%).
Disitamab vedotin 由一种人源化抗 HER-2 单克隆抗体组成,该单克隆抗体通过可切割的缬氨酸-瓜氨酸连接子偶联到 MMAE 有效载荷上 [ 150]。它于2021年6月在中国获得有条件批准,并在美国获得FDA的快速通道指定[151]。在1期剂量递增研究中,21例HER2过表达晚期实体癌患者每两周接受一次0.1mg/kg-2.5mg/kg的disitamab vedotin治疗[152]。最常见的治疗相关不良事件为白细胞减少(61.1%)、中性粒细胞减少(52.8%)、疲劳(50.0%)、麻木(44.4%)、AST升高(30.6%)和ALT升高(27.8%)。最大耐受剂量未确定至2.5mg / kg。在中国纳入的5项1-3期临床试验中,最常见的治疗相关不良事件是白细胞减少(55.4%)、脱发(54.6%)、中性粒细胞减少(50.6%)、天冬氨酸氨基转移酶升高(49.7%)、疲劳(46.3%)、丙氨酸转氨酶升高(42.9%)和感觉减退(40.9%)[151]。最常见的 3 级≥不良事件是中性粒细胞减少 (16.9%)、白细胞减少 (10.9%) 和感觉减退 (8.9%)。
Disitamab vedotin 由一种人源化抗 HER-2 单克隆抗体组成,该单克隆抗体通过可切割的缬氨酸-瓜氨酸连接子偶联到 MMAE 有效载荷上 [ 150]。它于2021年6月在中国获得有条件批准,并在美国获得FDA的快速通道指定[151]。在1期剂量递增研究中,21例HER2过表达晚期实体癌患者每两周接受一次0.1mg/kg-2.5mg/kg的disitamab vedotin治疗[152]。最常见的治疗相关不良事件为白细胞减少(61.1%)、中性粒细胞减少(52.8%)、疲劳(50.0%)、麻木(44.4%)、AST升高(30.6%)和ALT升高(27.8%)。最大耐受剂量未确定至2.5mg / kg。在中国纳入的5项1-3期临床试验中,最常见的治疗相关不良事件是白细胞减少(55.4%)、脱发(54.6%)、中性粒细胞减少(50.6%)、天冬氨酸氨基转移酶升高(49.7%)、疲劳(46.3%)、丙氨酸转氨酶升高(42.9%)和感觉减退(40.9%)[151]。最常见的 3 级≥不良事件是中性粒细胞减少 (16.9%)、白细胞减少 (10.9%) 和感觉减退 (8.9%)。
3.3. Frequently Reported ADC-Associated Dose-Limiting Toxicities
3.3. 经常报告的ADC相关剂量限制性毒性
3.3.1. Neutropenia 3.3.1. 中性粒细胞减少症
Neutrophils, which account for 40% to 70% of granulocytes, are an essential component of the innate immune system [153]. Neutrophils are produced in the bone marrow at a rapid rate (approximately 1011 cells/day) through the differentiation of hematopoietic stem cells. They have a relatively short blood circulation half-life (about one day) [154,155,156]. These unique characteristics render neutrophils more vulnerable to anti-neoplastic agents compared to other myeloid cells that have longer lifespans, including platelets (8 days) and erythrocytes (120 days) [18]. Interruption of hematopoietic cellular division in the bone marrow often results in a reduced production of neutrophils and leads to susceptibility to serious infections and infection sequela, including sepsis and febrile neutropenia. Severe neutropenia is a frequent dose-limiting toxicity associated with most MMAE-ADCs that utilize a valine-citrulline cleavable linker, including brentuximab vedotin, polatuzumab vedotin, enfortumab vedotin, and tisotumab vedotin. In addition, grade ≥ 3 neutropenia is also commonly reported for ADCs utilizing other payloads with cleavable linkers, such calicheamicin (gemtuzumab ozogamicin and inotuzumab ozogamicin), SN-38 (sacituzumab govitecan), extecan (trastuzumab deruxtecan), PBD (loncastuximab tesirine), DM4 (coltuximab ravtansine), and doucarmycine (trastuzumab duocarmazine). Neutropenia is less common in patients treated with ADCs utilizing stable linkers, including MMAF-conjugates (belantamab mafodotin) or DM1-conjugates (trastuzumab emtansine).
中性粒细胞占粒细胞的40%-70%,是先天免疫系统的重要组成部分[153]。中性粒细胞通过造血干细胞的分化在骨髓中以快速的速度(约 10 11 个细胞/天)产生。它们的血液循环半衰期相对较短(约一天)[154,155,156]。与其他寿命更长的髓系细胞(包括血小板(8日)和红细胞(120天))相比,这些独特的特征使中性粒细胞更容易受到抗肿瘤药物的影响[18]。骨髓中造血细胞分裂的中断通常会导致中性粒细胞的产生减少,并导致对严重感染和感染后遗症的易感性,包括脓毒症和发热性中性粒细胞减少症。重度中性粒细胞减少症是一种常见的剂量限制性毒性,与大多数使用缬氨酸-瓜氨酸可裂解接头的 MMAE-ADC 相关,包括 brentuximab vedotin、polatuzumab vedotin、enfortumab vedotin 和 tisotumab vedotin。此外,≥ 级中性粒细胞减少症也常见于使用其他具有可切割接头的有效载荷的 ADC,例如卡利米星(吉妥珠单抗奥佐米星和伊妥珠单抗奥佐米辛)、SN-38(戈维康沙妥珠单抗)、extecan(曲妥珠单抗 deruxtecan)、PBD(loncastuximab tesirine)、DM4(coltuximab ravtansine)和杜卡米辛(曲妥珠单抗 duocarmazine)。中性粒细胞减少症在接受使用稳定连接子的 ADC 治疗的患者中较少见,包括 MMAF 偶联物(belantamab mafodotin)或 DM1 偶联物(曲妥珠单抗 emtansine)。
中性粒细胞占粒细胞的40%-70%,是先天免疫系统的重要组成部分[153]。中性粒细胞通过造血干细胞的分化在骨髓中以快速的速度(约 10 11 个细胞/天)产生。它们的血液循环半衰期相对较短(约一天)[154,155,156]。与其他寿命更长的髓系细胞(包括血小板(8日)和红细胞(120天))相比,这些独特的特征使中性粒细胞更容易受到抗肿瘤药物的影响[18]。骨髓中造血细胞分裂的中断通常会导致中性粒细胞的产生减少,并导致对严重感染和感染后遗症的易感性,包括脓毒症和发热性中性粒细胞减少症。重度中性粒细胞减少症是一种常见的剂量限制性毒性,与大多数使用缬氨酸-瓜氨酸可裂解接头的 MMAE-ADC 相关,包括 brentuximab vedotin、polatuzumab vedotin、enfortumab vedotin 和 tisotumab vedotin。此外,≥ 级中性粒细胞减少症也常见于使用其他具有可切割接头的有效载荷的 ADC,例如卡利米星(吉妥珠单抗奥佐米星和伊妥珠单抗奥佐米辛)、SN-38(戈维康沙妥珠单抗)、extecan(曲妥珠单抗 deruxtecan)、PBD(loncastuximab tesirine)、DM4(coltuximab ravtansine)和杜卡米辛(曲妥珠单抗 duocarmazine)。中性粒细胞减少症在接受使用稳定连接子的 ADC 治疗的患者中较少见,包括 MMAF 偶联物(belantamab mafodotin)或 DM1 偶联物(曲妥珠单抗 emtansine)。
ADC-associated neutropenia appears to correlate with the cumulative plasma exposure to released payload (e.g., following premature release or following distribution from intracellular sites of ADC catabolism). Membrane-permeable payloads readily distribute into bone marrow and into differentiating hematopoietic cells [157]. The deconjugation of intact ADCs within extracellular fluid within the bone marrow compartment may also contribute to myelotoxicity, as differentiating neutrophils secrete serine proteases that can cleave ADC linkers [157]. In addition to the off-target mechanisms mentioned above, target-dependent mechanisms may also contribute to neutropenia for ADCs targeting leukemic antigens [77,158,159,160]. In a recent systemic analysis, Haubner et al. quantified the expression of leukemic stem cell makers. Several of them, including CD33, CD123, and CLL-1, exhibited a high expression level not only in leukemic stem cells but also in normal hematopoietic stem/progenitor cells, which are precursors for neutrophil production [161].
ADC相关的中性粒细胞减少似乎与释放有效载荷的累积血浆暴露相关(例如,过早释放后或ADC分解代谢从细胞内位点分布后)。膜渗透性有效载荷很容易分布到骨髓和分化造血细胞中[157]。骨髓区室内细胞外液中完整ADC的解偶联也可能导致骨髓毒性,因为分化中性粒细胞分泌丝氨酸蛋白酶,可以切割ADC接头[157]。除了上述脱靶机制外,靶点依赖性机制还可能导致靶向白血病抗原的ADC的中性粒细胞减少[77,158,159,160]。在最近的系统分析中,Haubner等人量化了白血病干细胞制造商的表达。其中几种,包括CD33、CD123和CLL-1,不仅在白血病干细胞中表现出高表达水平,而且在正常造血干细胞/祖细胞中也表现出高表达水平,这些细胞是中性粒细胞产生的前体[161]。
ADC相关的中性粒细胞减少似乎与释放有效载荷的累积血浆暴露相关(例如,过早释放后或ADC分解代谢从细胞内位点分布后)。膜渗透性有效载荷很容易分布到骨髓和分化造血细胞中[157]。骨髓区室内细胞外液中完整ADC的解偶联也可能导致骨髓毒性,因为分化中性粒细胞分泌丝氨酸蛋白酶,可以切割ADC接头[157]。除了上述脱靶机制外,靶点依赖性机制还可能导致靶向白血病抗原的ADC的中性粒细胞减少[77,158,159,160]。在最近的系统分析中,Haubner等人量化了白血病干细胞制造商的表达。其中几种,包括CD33、CD123和CLL-1,不仅在白血病干细胞中表现出高表达水平,而且在正常造血干细胞/祖细胞中也表现出高表达水平,这些细胞是中性粒细胞产生的前体[161]。
3.3.2. Thrombocytopenia 3.3.2. 血小板减少症
Thrombocytopenia is a common off-target toxicity associated with ADCs that utilize stable linkers (SMCC-DM1 or mc-MMAF) and also for the potent DNA-crosslinking payload calicheamicin. For example, grade ≥ 3 thrombocytopenia occurred in 99% of acute myeloid leukemia patients [80] and 42% of acute lymphoblastic leukemia patients treated with gemtuzumab ozogamicin [98]. Grade ≥ 3 thrombocytopenia was reported in 14.5% of breast cancer patients treated with trastuzumab emtansine [66] and in 21% of multiple myeloma patients treated with belantamab mafodotin [119].
血小板减少症是一种常见的脱靶毒性,与利用稳定接头(SMCC-DM1 或 mc-MMAF)的 ADC 以及有效的 DNA 交联有效载荷卡利霍霉素有关。例如,99%的急性髓系白血病患者[80]和42%的吉妥珠单抗奥佐米星治疗的急性淋巴细胞白血病患者发生≥级3级血小板减少症[98]。据报道,14.5%的乳腺癌患者接受曲妥珠单抗emtansine治疗[66],21%接受belantamab mafodotin治疗的多发性骨髓瘤患者出现≥级血小板减少症[119]。
血小板减少症是一种常见的脱靶毒性,与利用稳定接头(SMCC-DM1 或 mc-MMAF)的 ADC 以及有效的 DNA 交联有效载荷卡利霍霉素有关。例如,99%的急性髓系白血病患者[80]和42%的吉妥珠单抗奥佐米星治疗的急性淋巴细胞白血病患者发生≥级3级血小板减少症[98]。据报道,14.5%的乳腺癌患者接受曲妥珠单抗emtansine治疗[66],21%接受belantamab mafodotin治疗的多发性骨髓瘤患者出现≥级血小板减少症[119]。
The definitive mechanism of thrombocytopenia remains elusive. The non-specific uptake of ADCs in differentiating megakaryocytes via either FcγR-mediated or micropinocytosis pathways was proposed as a probable cause [42,43,44]. However, based on the observation that the clinical manifestation of ADC-associated thrombocytopenia usually occurs within 24 h, which is significantly shorter than the typical platelet lifespan of 8 to 10 days, additional factors might be contributing to thrombocytopenia. Indeed, Guffroy et al. showed that monkeys treated with a calicheamicin-based ADC exhibited significant damage in the sinusoidal epithelial cells and pronounced sequestration and accumulation of platelets in the hepatic sinusoids three days after treatment, which coincided with the nadir of platelet reduction [58].
血小板减少症的明确机制仍然难以捉摸。ADCs通过FcγR介导或微胞饮作用途径分化巨核细胞的非特异性摄取被认为是可能的原因[42,43,44]。然而,根据观察,ADC相关血小板减少症的临床表现通常发生在24小时内,明显短于典型的血小板寿命8至10天,其他因素可能导致血小板减少症。事实上,Guffroy等人发现,用基于卡利霉素的ADC治疗的猴子在治疗后3天表现出明显的肝窦上皮细胞损伤,肝窦中血小板明显隔离和积聚,这与血小板减少的最低点相吻合[58]。
血小板减少症的明确机制仍然难以捉摸。ADCs通过FcγR介导或微胞饮作用途径分化巨核细胞的非特异性摄取被认为是可能的原因[42,43,44]。然而,根据观察,ADC相关血小板减少症的临床表现通常发生在24小时内,明显短于典型的血小板寿命8至10天,其他因素可能导致血小板减少症。事实上,Guffroy等人发现,用基于卡利霉素的ADC治疗的猴子在治疗后3天表现出明显的肝窦上皮细胞损伤,肝窦中血小板明显隔离和积聚,这与血小板减少的最低点相吻合[58]。
3.3.3. Peripheral Neuropathy
3.3.3. 周围神经病变
Clinical manifestation of peripheral neuropathy includes sensory-related symptoms, such as numbness, tingling, and pain in the extremities, or, to a lesser extent, motor-related symptoms such as muscle weakness [18]. Peripheral neuropathy is a common adverse effect associated with tubulin-inhibiting chemotherapeutic agents such as taxanes and vinca alkaloids [162]. Similarly, peripheral neuropathy is a significant dose-limiting off-target toxicity associated with ADCs utilizing tubulin inhibitor payloads combined with cleavable linkers such as vc-MMAE, SPP-DM1, and SPDB-DM4 [102,134,163,164]. The mechanism behind peripheral neuropathy is hypothesized to be peripheral axonopathy induced by free payload released in the systemic circulation [18]. Even though peripheral neurons are not actively proliferating, a functional microtubule is critical for protein transport from the cell body to the distal synapses. Therefore, the disruption of microtubules by tubulin-inhibiting payloads may result in peripheral neuropathy [18].
周围神经病变的临床表现包括感觉相关症状,如四肢麻木、刺痛和疼痛,或较小程度的运动相关症状,如肌无力[18]。周围神经病变是一种常见的不良反应,与微管蛋白抑制化疗药物(如紫杉烷类和长春花生物碱)有关[162]。类似地,周围神经病变是一种显著的剂量限制性脱靶毒性,与利用微管蛋白抑制剂有效载荷与可切割接头(如vc-MMAE、SPP-DM1和SPDB-DM4)结合的ADC相关[102,134,163,164]。周围神经病变背后的机制被推测为由体循环中释放的游离有效载荷诱发的外周轴突病[18]。尽管外周神经元没有积极增殖,但功能性微管对于蛋白质从细胞体到远端突触的运输至关重要。因此,微管蛋白抑制有效载荷对微管的破坏可能导致周围神经病变[18]。
周围神经病变的临床表现包括感觉相关症状,如四肢麻木、刺痛和疼痛,或较小程度的运动相关症状,如肌无力[18]。周围神经病变是一种常见的不良反应,与微管蛋白抑制化疗药物(如紫杉烷类和长春花生物碱)有关[162]。类似地,周围神经病变是一种显著的剂量限制性脱靶毒性,与利用微管蛋白抑制剂有效载荷与可切割接头(如vc-MMAE、SPP-DM1和SPDB-DM4)结合的ADC相关[102,134,163,164]。周围神经病变背后的机制被推测为由体循环中释放的游离有效载荷诱发的外周轴突病[18]。尽管外周神经元没有积极增殖,但功能性微管对于蛋白质从细胞体到远端突触的运输至关重要。因此,微管蛋白抑制有效载荷对微管的破坏可能导致周围神经病变[18]。
3.3.4. Ocular Toxicity 3.3.4. 眼毒性
Ocular toxicity is one of the key off-target dose-limiting toxicities of ADCs containing the SPDB-DM4 linker-payload, such as cantuzumab ravtansine [165], mirvetuximab soravtansine [166], and coltuximab ravtansine [167], or the mc-MMAF linker-payload, such as belantamab mafodotin [119], AGS-16C3F [168], and SGN-75 [169]. The susceptibility of the eye to ADC cytotoxicity is possibly due to several factors, including an ample blood supply, the presence of rapidly dividing epithelial cell populations, and the high expression of several cell-surface receptors [170]. In clinical settings, symptoms of ADC-associated ocular adverse events include blurred vision, keratitis, dry eye, and microcystic epithelial damage. The mechanism underlying ocular toxicity associated with ADCs is still poorly understood. The non-specific uptake of intact ADCs via macropinocytosis was suggested by Zhao et al. as the mechanism behind the off-target ocular toxicity of ADCs [40]. However, the target-mediated uptake of ADC might also be relevant [171]. Table 2 summarizes the commonly reported dose-limiting toxicities associated with ADC treatments.
眼毒性是含有 SPDB-DM4 接头有效载荷的 ADC 的关键脱靶剂量限制性毒性之一,例如坎妥珠单抗拉夫坦辛 [ 165]、米维妥昔单抗 soravtansine [ 166] 和科妥昔单抗拉夫坦辛 [ 167],或 mc-MMAF 接头有效载荷,例如 belantamab mafodotin [ 119]、AGS-16C3F [ 168] 和 SGN-75 [ 169]。眼睛对ADC细胞毒性的易感性可能由多种因素引起,包括充足的血液供应、快速分裂的上皮细胞群的存在以及几种细胞表面受体的高表达[170]。在临床环境中,ADC 相关眼部不良事件的症状包括视力模糊、角膜炎、干眼症和微囊性上皮损伤。与ADC相关的眼毒性机制仍知之甚少。Zhao等人认为,通过巨胞饮作用非特异性摄取完整ADC是ADC脱靶眼毒性背后的机制[40]。然而,靶向介导的ADC摄取也可能相关[171]。表2总结了与ADC治疗相关的常见剂量限制性毒性。
眼毒性是含有 SPDB-DM4 接头有效载荷的 ADC 的关键脱靶剂量限制性毒性之一,例如坎妥珠单抗拉夫坦辛 [ 165]、米维妥昔单抗 soravtansine [ 166] 和科妥昔单抗拉夫坦辛 [ 167],或 mc-MMAF 接头有效载荷,例如 belantamab mafodotin [ 119]、AGS-16C3F [ 168] 和 SGN-75 [ 169]。眼睛对ADC细胞毒性的易感性可能由多种因素引起,包括充足的血液供应、快速分裂的上皮细胞群的存在以及几种细胞表面受体的高表达[170]。在临床环境中,ADC 相关眼部不良事件的症状包括视力模糊、角膜炎、干眼症和微囊性上皮损伤。与ADC相关的眼毒性机制仍知之甚少。Zhao等人认为,通过巨胞饮作用非特异性摄取完整ADC是ADC脱靶眼毒性背后的机制[40]。然而,靶向介导的ADC摄取也可能相关[171]。表2总结了与ADC治疗相关的常见剂量限制性毒性。
4. Strategies to Reduce Toxicities
4. 减少毒性的策略
The high attrition rate of ADCs containing antimitotic payloads is primarily due to a suboptimal efficacy at the MTD during clinical trials. In response, the recent development of ADCs has shown increased utilization of more potent DNA-damaging payloads such as PBD dimers, duocarmycins, and indolinobenzodiazepine dimers [24,28,172]. Unfortunately, even though these highly potent ADCs often lead to a higher efficacy, their clinical success has been limited due to the increased incidence of life-threatening toxicities. For instance, treatment with vadastuximab talirine, an anti-CD33 PBD-ADC developed by Seattle Genetics, reached a 70% complete remission rate in AML patients; however, due to the occurrence of several treatment-related fatal events, the clinical development of vadastuximab talirine has been terminated [28]. A recent analysis by Saber and Leighton from the FDA found that 47% of investigational new drug applications for PBD-conjugated ADCs between 2013 and 2017 were discontinued, mainly due to safety concerns [22]. These outcomes suggest that unwanted toxicity restricts the clinical benefit of ADCs by limiting the tolerable doses to amounts below the levels required to provide meaningful or optimal anti-cancer efficacy. In a formal comparison of effective exposure levels identified in preclinical studies with exposure profiles at the maximum tolerated ADC doses in the clinic, de Goeij and Lambert concluded that, for many ADCs, the exposures at the clinical MTDs were significantly lower than the exposures required for efficacy in preclinical models [172]. Much focus in the ADC field is shifting to strategies to widen the therapeutic window through the mitigation of ADC toxicity, as this approach, if successful, might enable increased MTDs and subsequently improved clinical outcomes. This section will discuss several strategies that have demonstrated improvements in ADC therapeutic windows and/or reductions in ADC toxicity in preclinical models or clinical settings.
含有抗有丝分裂有效载荷的ADC的高损耗率主要是由于临床试验期间MTD的疗效欠佳。作为回应,ADCs的最新发展表明,对更有效的DNA损伤有效载荷的利用率增加,如PBD二聚体、duocarmycins和吲哚啉并二氮卓二聚体[24,28,172]。不幸的是,尽管这些高效ADC通常能带来更高的疗效,但由于危及生命的毒性发生率增加,它们的临床成功率有限。例如,Seattle Genetics 开发的抗 CD33 PBD-ADC vadastuximab talirine 治疗在 AML 患者中达到了 70% 的完全缓解率;然而,由于发生了几起与治疗相关的致死事件,伐达昔单抗塔利林的临床开发已经终止[28]。FDA的Saber和Leighton最近的一项分析发现,2013年至2017年间,PBD偶联ADC的研究性新药申请中有47%被终止,主要是出于安全考虑[22]。这些结果表明,不需要的毒性限制了ADC的临床益处,将可耐受剂量限制在低于提供有意义或最佳抗癌疗效所需的水平的水平。在将临床前研究中确定的有效暴露水平与临床中最大耐受ADC剂量的暴露曲线进行正式比较时,de Goeij和Lambert得出结论,对于许多ADC,临床MTD的暴露量明显低于临床前模型中疗效所需的暴露量[172]。 ADC领域的许多焦点正在转向通过减轻ADC毒性来扩大治疗窗口的策略,因为这种方法如果成功,可能会增加MTD并随后改善临床结果。本节将讨论几种策略,这些策略已证明在临床前模型或临床环境中改善了ADC治疗窗口和/或降低了ADC毒性。
含有抗有丝分裂有效载荷的ADC的高损耗率主要是由于临床试验期间MTD的疗效欠佳。作为回应,ADCs的最新发展表明,对更有效的DNA损伤有效载荷的利用率增加,如PBD二聚体、duocarmycins和吲哚啉并二氮卓二聚体[24,28,172]。不幸的是,尽管这些高效ADC通常能带来更高的疗效,但由于危及生命的毒性发生率增加,它们的临床成功率有限。例如,Seattle Genetics 开发的抗 CD33 PBD-ADC vadastuximab talirine 治疗在 AML 患者中达到了 70% 的完全缓解率;然而,由于发生了几起与治疗相关的致死事件,伐达昔单抗塔利林的临床开发已经终止[28]。FDA的Saber和Leighton最近的一项分析发现,2013年至2017年间,PBD偶联ADC的研究性新药申请中有47%被终止,主要是出于安全考虑[22]。这些结果表明,不需要的毒性限制了ADC的临床益处,将可耐受剂量限制在低于提供有意义或最佳抗癌疗效所需的水平的水平。在将临床前研究中确定的有效暴露水平与临床中最大耐受ADC剂量的暴露曲线进行正式比较时,de Goeij和Lambert得出结论,对于许多ADC,临床MTD的暴露量明显低于临床前模型中疗效所需的暴露量[172]。 ADC领域的许多焦点正在转向通过减轻ADC毒性来扩大治疗窗口的策略,因为这种方法如果成功,可能会增加MTD并随后改善临床结果。本节将讨论几种策略,这些策略已证明在临床前模型或临床环境中改善了ADC治疗窗口和/或降低了ADC毒性。
4.1. Modifying Conjugation Technology or Drug/Linker Chemistry
4.1. 修改偶联技术或药物/接头化学
The conventional conjugation of methods employing non-specific linkage of amine-based lysine or thiol-based cysteine residues of the antibody leads to considerable heterogeneity in the DAR and hydrophobicity [173]. The degree of drug loading ranges from no loading (DAR = 0) to highly-loaded ADCs (DAR ≥ 6). Highly-loaded ADCs are often unstable in plasma and exhibit high rates of non-specific uptake in the liver, leading to increased off-target toxicity [36,174]. Several approaches have been developed to precisely conjugate a designated number of cytotoxic payloads at defined antibody locations. These technologies would enable a more precise control of DAR and improve the stability of the conjugated payload due to its conjugation to more stable site, while also simplifying the in-process control and ADC manufacturing [175]. The first site-specific conjugation strategy was developed by Junutula et al. and allows the thiol-based linkage of payloads to extra cysteine residues introduced to the antibody at the Ala114 position of the CH1 domain via site-directed mutagenesis [176]. The engineered antibody, called THIOMAB, enables the site-specific coupling of MMAE payloads via a thiol-reactive linker to form THIOMAB-drug conjugates (TDCs). For instance, an anti-MUC16 TDC exhibited a similar in vivo efficacy to ADC, despite having 2-fold lower drug loading. Safety studies demonstrated that healthy rats tolerated TDC at a much higher dose (4150 µg/m2 MMAE, or 100 mg/kg TDC) than ADC (2250 µg/m2 MMAE, or 25 mg/kg ADC). In addition, reduced hematologic and hepatic toxicities were observed in rats and monkeys treated with TDC compared to ADC [176]. Since the inception of the THIOMAB technology, numerous site-specific conjugation methods have emerged, and several have reached clinical development [24,177,178]. More extensive reviews of these methods are discussed elsewhere [179,180,181,182,183,184].
采用胺基赖氨酸或硫醇基半胱氨酸残基的非特异性键的方法的常规偶联导致DAR和疏水性存在相当大的异质性[173]。药物载量范围从无载荷(DAR = 0)到高载ADC(DAR ≥ 6)。高负荷的ADC在血浆中通常不稳定,在肝脏中表现出较高的非特异性摄取率,导致脱靶毒性增加[36,174]。已经开发了几种方法,可以在确定的抗体位置精确偶联指定数量的细胞毒性有效载荷。这些技术将能够更精确地控制DAR,并提高共轭有效载荷的稳定性,因为它与更稳定的位点共轭,同时也简化了过程控制和ADC制造[175]。Junutula等人开发了第一种位点特异性偶联策略,允许通过定点诱变将有效载荷与CH1结构域Ala114位置引入抗体的额外半胱氨酸残基进行基于硫醇的连接[176]。这种名为 THIOMAB 的工程抗体能够通过巯醇反应性接头对 MMAE 有效载荷进行位点特异性偶联,以形成 THIOMAB 药物偶联物 (TDC)。例如,抗MUC16 TDC表现出与ADC相似的体内功效,尽管其载药量低2倍。安全性研究表明,健康大鼠耐受的TDC剂量(4150μg/m 2 MMAE,或100mg / kg TDC)远高于ADC(2250μg/m 2 MMAE,或25mg / kg ADC)。此外,与ADC相比,TDC治疗的大鼠和猴子的血液学和肝脏毒性降低[176]。 自硫代单抗技术问世以来,已经出现了许多位点特异性偶联方法,其中一些方法已进入临床开发阶段[24,177,178]。这些方法的更广泛综述详见其他专题[179, 180, 181, 182, 183, 184]。
采用胺基赖氨酸或硫醇基半胱氨酸残基的非特异性键的方法的常规偶联导致DAR和疏水性存在相当大的异质性[173]。药物载量范围从无载荷(DAR = 0)到高载ADC(DAR ≥ 6)。高负荷的ADC在血浆中通常不稳定,在肝脏中表现出较高的非特异性摄取率,导致脱靶毒性增加[36,174]。已经开发了几种方法,可以在确定的抗体位置精确偶联指定数量的细胞毒性有效载荷。这些技术将能够更精确地控制DAR,并提高共轭有效载荷的稳定性,因为它与更稳定的位点共轭,同时也简化了过程控制和ADC制造[175]。Junutula等人开发了第一种位点特异性偶联策略,允许通过定点诱变将有效载荷与CH1结构域Ala114位置引入抗体的额外半胱氨酸残基进行基于硫醇的连接[176]。这种名为 THIOMAB 的工程抗体能够通过巯醇反应性接头对 MMAE 有效载荷进行位点特异性偶联,以形成 THIOMAB 药物偶联物 (TDC)。例如,抗MUC16 TDC表现出与ADC相似的体内功效,尽管其载药量低2倍。安全性研究表明,健康大鼠耐受的TDC剂量(4150μg/m 2 MMAE,或100mg / kg TDC)远高于ADC(2250μg/m 2 MMAE,或25mg / kg ADC)。此外,与ADC相比,TDC治疗的大鼠和猴子的血液学和肝脏毒性降低[176]。 自硫代单抗技术问世以来,已经出现了许多位点特异性偶联方法,其中一些方法已进入临床开发阶段[24,177,178]。这些方法的更广泛综述详见其他专题[179, 180, 181, 182, 183, 184]。
In addition to site-specific conjugation, the modification of the linker can also reduce ADC hydrophobicity and reduce toxicity. Burke and Simmons et al. demonstrated that PEGylation of the drug-linker component increased the hydrophilicity of ADCs, which led to improved PK, tolerability, and efficacy [185,186]. They incorporated a PEG polymer with varying lengths from 2 to 24 into the ADC linker. Then, the impact of the spacer on the ADC’s in vitro potency and in vivo efficacy/tolerability was evaluated. While all ADCs with a PEG spacer had similar in vitro cellular cytotoxicity, ADCs with a PEG spacer with a length of eight or greater had slower clearance and superior in vivo anti-tumor activity. Notably, the tolerability of the ADC was significantly improved with the addition of the PEG spacer [185]. At the 50 mg/kg dose, all animals treated with non-PEGylated ADC lost more than 20% of body weight on day 6, while the animal treated with PEG12-ADC maintained minimal change in body weight. Further studies showed that ADCs with PEGylated linkers had reduced peak plasma and tissue concentrations, leading to substantially less hematologic toxicity [186]. In addition to MMAE, this PEGylation approach have been utilized for other payloads including maytansinoid [187,188,189], duocamycin [148], MTA eribulin [190], and PBD [144,191,192,193,194]. Several of these ADCs are currently being investigated in clinical trials, and two ADCs with PEGylated linkers, loncastuximab tesirine and sacituzumab govitecan, have been FDA-approved for clinical use [143,195].
The advancement of self-immolative linker chemistry has also improved the stability and tolerability of ADCs utilizing cleavable linkers. In the early 1980s, the para-aminobenzyloxycarbonyl (PABC) self-immolate linker chemistry was developed by Carl et al. for use in prodrug design [196]. Since then, the PABC linker type has been extensively utilized in ADC linker technology, particularly for MMAE-conjugated ADC. Upon cleavage by lysosomal enzymes after ADC internalization into the target cells, the PABC spacer in the vc-PAB-MMAE linker/payload goes through a cascade of disassembly reactions and facilitates the release of untethered MMAE intracellularly [197]. The vc-PABC linker exhibits superior plasma stability and improved toxicity profile compared to the earlier cleavable linker generations, such as the pH-sensitive hydrazone linker used in calicheamicin-based ADCs or disulfide reducible linker used in DM1-based ADCs [198]. This linker technology has achieved significant success with its application in four MMAE-ADCs out of the twelve approved ADCs. In addition, the PABC spacer also has been used in the development of PBD-based ADCs, including the approved ADC loncastuximab tesirine. Recently, Pillow et al. have developed sophisticated self-immolative spacer chemistry that enabled direct conjugation of a cytotoxic payload to engineered cysteine residues [30]. This spacer facilitates a stable disulfide bridge, leading to enhanced plasma PK while allowing a rapid traceless release of free payloads intracellularly. This technology improved the tolerability of PBD-ADCs compared to the conventional peptide-PABC linker technology [199]. In this study, both the conventional peptide-ADC and the self-immolative disulfide-ADC displayed similar in vivo potency. However, while the self-immolative ADC was well-tolerated up to 10 mg/kg dose in rats, the animals treated with a 5 mg/kg dose of the peptide-ADC experienced significant weight loss.
Previous generations of cleavable peptide linkers, including the vc linker widely used in MMAE ADC technology, are susceptible to the peptidases present in plasma and in tissues. Recent developments in peptide-based linker technology have generated several linkers with appreciably enhanced stability against non-specific peptidase cleavage. For example, Chuprakov et al. introduced a β-glucuronide moiety to sterically protect the vc linker from non-specific peptidase cleavage in the systemic circulation [200]. Utilizing this technology, the authors developed an anti-CD79b ADC with a tandem-cleavage linker, which requires two sequential enzymatic cleavage events to release the cytotoxic payload: (1) removal of the monosaccharide group by β-glucuronidase present in the lysosomal compartment and, subsequently, (2) cleavage of the vc linkage by lysosomal peptidases. Compared to the anti-CD79b ADCs with a “standard” mono-cleavage vc-linker, the ADC with the tandem-cleavage linker technology demonstrated marked improvement in plasma stability and in vivo efficacy. In a tolerability study in rats, the animals treated with the mono-cleavage ADCs exhibited a significant reduction in circulating white blood cells, including monocytes, neutrophils, and eosinophils, whereas animals treated with the tandem-cleavage ADC exhibited minimal myelotoxicity. In another example, the Tsuchikama group developed tripeptide linkers with enhanced resistance to plasma and tissue-specific peptidases [201,202]. In a recent published study, they have optimized glutamic acid-glycine-citrulline (EGCit) linkers that have significantly improved resistance to neutrophil elastase compared to the conventional vc linker [202]. In an in vitro cytotoxicity study, treatment with an anti-HER2 EGCit-ADC had a minimal impact on cell viability of CD15+/CD66b+ differentiating human neutrophils, whereas treatment with an anti-HER2 ADC employing glutamic acid-valine-citrulline linker led to ~50% reduction in cell viability. In an in vivo toxicology study, mice treated with 80 mg/kg of the anti-HER2 EGCit-ADC exhibited minimal weight loss, whereas treatment with the same dose of an anti-HER2 ADC utilizing the conventional vc-linker led to ~15% weight loss. In addition, significant liver damage, as indicated by increased alanine aminotransferase (ALT), increased aspartate aminotransferase (AST), increased alkaline phosphatase (ALKP), and decreased blood urea nitrogen (BUN), was observed in the animals treated with 80 mg/kg of trastuzumab deruxtecan or trastuzumab emtansine, but not in animals treated with the same dose of the EGCit-ADC.
The modification of the chemical structure of the payload may also lead to an improved ADC tolerability. For example, Miller et al. permutated the chemical structure of the DNA-crosslinking PBD-dimer to yield novel DNA-alkylating metabolites, indolinobenzodiazepine dimers (IGNs), through a reduction in one of the DNA-reactive imine functional groups [203]. IGN-conjugated ADCs exhibited an enhanced bystander effect (relative to PBD-conjugated ADCs), due to a more efficient release of free payload from the DNA adducts [204], which translated into better in vivo efficacy in murine tumor models [205]. In addition, mice tolerated IGN-conjugated ADCs at a 6 mg/kg dose compared to a 2.8 mg/kg dose for PBD-conjugated ADCs. At a 6 mg/kg dose, all of the mice treated with PBD-ADC (n = 8) experienced prolonged body weight loss and were dead by 60 days after treatment.
4.2. Antibody Modifications
Since most of the targets for ADCs also have some level of expression in healthy tissue [206], the delivery of the cytotoxic payload in these tissues through ADC binding can lead to on-target off-site toxicity. To reduce on-target toxicities, probody drug conjugates (PDC) have been developed where binding regions of the parent antibody are masked with anti-idiotypic peptides, which connect to the N-terminus of the light chain via a protease-susceptible peptide linker [207]. The intent is to enhance tumor selectivity through preferential activation within the tumor microenvironment, where upregulated protease activity [208,209] cleaves the masking peptide, thus enabling PDC binding to the target antigen. Slower cleavage of the masking peptide reduces the extent of PDC binding to the target antigen in healthy cells, and reduces on-target, off-site toxicity. In addition to potentially enabling selective activation within the tumor environment, PDC constructs, and complexes of ADC with anti-idiotypic binding domains, may enhance the ADC’s intra-tumoral distribution, efficacy, and therapeutic index by decreasing the impact of the “binding site barrier” within solid tumors [210,211,212,213]. In one notable preclinical example, Sagert et al. showed that a rodent Jagged antigen-binding PDC demonstrated substantially reduced weight loss relative to an ADC developed with the parent mAb, while the ADC and PDC produced similar efficacy [214]. Several PDC are under preclinical or clinical development [215,216,217,218].
Another approach to reducing on-target toxicity is through the development of bispecific antibodies targeting two tumor-associated antigens. This approach enables enhanced selectivity because avid ADC binding is only observed on cells with high co-expression of both antigens [219,220]. Indeed, work by Mazor et al. demonstrated a significant increase the in vitro binding selectivity of anti-HER2/EGFR bispecific antibodies to HER2+/EGFR+ cells than either HER2+/EGFR- or HER2-/EGFR+ cells [221]. A consistent finding was also observed with an anti-CD4/CD70 bispecific antibody. In addition, they also showed that binding selectivity could be further improved in bispecific antibodies with low-affinity variants of each binding arm, which exhibit poor binding to cells with single target expression [222,223]. Sellmann et al. demonstrated the feasibility of this dual-targeting concept in ADC technology with an MMAE-conjugated anti-EGFR/c-Met bispecific ADC [224]. EGFR is expressed widely, and skin toxicity is a significant concern with EGFR targeting therapies [225,226]. Compared to the anti-EGFR MMAE-conjugated cetuximab, the bispecific ADC displayed reduced in vitro binding activity and reduced cellular cytotoxicity when applied to healthy cells (e.g., HepG2 and human keratinocytes), while demonstrating high potency in tumor cells that express both targets (EGFR and c-Met) [224]. Several additional bispecific ADCs have been recently reported, including anti-HER2/integrin MMAF-conjugated bispecific ADCs [227], an anti-HER2/CD63 bispecific ADC [228], and an anti-HER2/PRLR bispecific ADC [229].
4.3. Modifying Dosage Regimens
Modifying clinical dosing schedules through fractional dosing has proven to improve the therapeutic index of ADCs in clinical settings. For instance, gemtuzumab ozogamicin was initially approved in 2001 to treat CD33-positive AML at the dose of 9 mg/m2 given at least two weeks apart. However, a high incidence of severe clinical toxicities was observed, including thrombocytopenia (99%), neutropenia (97%), grade ≥ 3 hyperbilirubinemia (23%), and grade ≥ 3 elevated alanine transaminase or aspartate transaminase levels, while no significant improvements in efficacy were shown compared to the standard of care [80]. Consequently, gemtuzumab ozogamicin was withdrawn from the market in 2010. Recently, fractionated dosing of the ADC was introduced such that patients receive the dose of 9 mg/m2 over three doses of 3 mg/m2 on the 1st, then 4th, and the 7th days of the dosing cycle, resulting in a more favorable toxicity profile [84]. PK/PD analysis of gemtuzumab ozogamicin indicates that its toxicities are driven by peak plasma concentrations, while its efficacy is driven by exposure [8,230]. Therefore, fractionated dosing would decrease the peak plasma concentrations and reduce toxicities while the maintaining drug exposure levels. With the modified dosing regimen, the ADC was re-approved for clinical use in 2017.
4.4. Inverse Targeting Strategy
The Balthasar laboratory has recently introduced the use of payload binding antibodies for “inverse targeting” to improve the therapeutic selectivity of ADCs. Briefly, payload binding antibody fragments (a.k.a. payload binding selectivity enhancers or PBSE) are co-administered with ADCs to enable the binding, neutralization, and clearance of released payload from plasma, decreasing the exposure of released payload in tissues associated with ADC toxicities. Antibody fragments have been utilized extensively to mitigate the adverse effects of drugs and toxins [231,232], including the use of anti-drug antibodies to enhance the pharmacokinetic selectivity of regional chemotherapy [233,234,235]. For example, the subcutaneous administration of anti-methotrexate Fab fragments significantly increased the maximum tolerated dose of intraperitoneal methotrexate by more than 5-fold (from 1.9 mg/kg to 10 mg/kg) in mice bearing peritoneal tumors [236]. Additionally, intravenous anti-topotecan antibody therapy allowed a significant increase in the MTD of intraperitoneal topotecan in a mouse model of peritoneal cancer [237]. The application of PBSE for the inverse targeting of ADCs has been recently pursued by the Balthasar group as a means of enhancing the safety and therapeutic index of ADCs. ABC3315, an MMAE payload binding selectivity enhancer (PBSE) Fab fragment, selectively binds to free MMAE but not to ADC-conjugated MMAE [238]. In in vitro cell culture, the co-incubation of ABC3315 with polatuzumab vedotin or trastuzumab-vc-MMAE led to an increase in the selectivity ratio of the ADC potency/payload potency by 738-fold in Ramos lymphoma cells and by 385-fold in SKBR3 breast cancer cells. In an in vivo lymphoma animal model, the co-treatment of polatuzumab vedotin with ABC3315 did not reduce the anti-tumor efficacy of the ADC; however, the co-dosing of ABC3315 with 120 mg/kg polatuzumab vedotin at a 3-fold molar ratio relative to MMAE significantly decreased the nadir body weight loss from 11.86% (for ADC plus PBS treated group) to 4.13% (for ADC plus ABC3315 treated group) in Swiss-Webster mice. Similarly, an anti-DM4 PBSE single-domain antibody, DMOH9, was developed for the inverse targeting of DM4-conjugated ADCs [239]. Co-dosing DMOH9 with 55 mg/kg 7E7-DM4, an anti-CD123-SPDB-DM4 ADC, at a 10-fold molar ratio relative to DM4 significantly decreased the nadir body weight loss from 11.86% (for ADC plus PBS treated group) to 4.13% (for ADC plus DMOH9 treated group) in Swiss-Webster mice. In an AML mouse model, the co-treatment of 7E7-DM4 with DMOH9 did not reduce the efficacy of the ADC or the survival of the tumor-bearing mice compared to the treatment with 7E7-DM4 alone at 1 or 10 mg/kg. Administration of the ADC (alone) at 100 mg/kg led to rapid death in 80% of treated mice; however, co-administration of the PBSE enabled all mice to tolerate this dose of the ADC, and allowed dramatically enhanced anti-tumor activity and animal survival. The PBSE technology may have utility in mitigating many off-site toxicities mediated by released payload, potentially leading to substantially improved ADC safety and efficacy.
5. Conclusions
By targeting the differential expression of cell-surface antigens on cancer cells vs. healthy cells, antibody-drug conjugates aim to increase the selectivity of the delivery of anti-cancer agents, and to widen the therapeutic window of traditional chemotherapy. However, the clinical utility of ADCs is limited by dose-limiting off-site toxicities. Significant research efforts have been invested to understand ADC-mediated off-site toxicity, and to develop strategies to improve ADC tolerability and, thus, efficacy.
Author Contributions
Writing—original draft preparation, T.D.N.; Visualization, T.D.N.; Writing—review and editing, T.D.N., B.M.B. and J.P.B. All authors have read and agreed to the published version of the manuscript.
Funding
Research reported in this publication was funded by the National Cancer Institute (CA246785, CA256928) and by the University at Buffalo Center for Protein Therapeutics.
Conflicts of Interest
J.P.B. serves as the Director of the University at Buffalo Center for Protein Therapeutics, which is supported by an industry consortium. Through the consortium, J.P.B. has received research support from Abbvie, Amgen, Astra Zeneca, CSL-Behring, Eli Lilly, Genentech, GSK, Janssen, Merck, Roche, and Sanofi. During the course of this work, J.P.B. has received consulting fees from companies involved with the development of cancer therapies, including Abbvie, Amgen, Eli Lilly, and Merck. J.P.B., B.M.B, and T.D.N. are founders of Abceutics, Inc., which is pursuing the development of payload binding agents to enhance the therapeutic selectivity of antibody-drug conjugates. J.P.B, B.M.B. and T.D.N. have financial interest through WO2021113740A1.
References
- Chau, C.H.; Steeg, P.S.; Figg, W.D. Antibody–drug conjugates for cancer. Lancet 2019, 394, 793–804. [Google Scholar] [CrossRef] [PubMed]
- Mecklenburg, L. A Brief Introduction to Antibody–Drug Conjugates for Toxicologic Pathologists. Toxicol. Pathol. 2018, 46, 746–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pysz, I.; Jackson, P.J.M.; Barlow, D.J.; Rahman, K.M.; Thurston, D.E. UPLC-based assay to assess the hydrophobicity of Antibody-Drug Conjugate (ADC) payloads. J. Chromatogr. B 2020, 1146, 122075. [Google Scholar] [CrossRef]
- Lobo, E.D.; Hansen, R.J.; Balthasar, J.P. Antibody pharmacokinetics and pharmacodynamics. J. Pharm. Sci. 2004, 93, 2645–2668. [Google Scholar] [CrossRef]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clin. Pharmacol. Ther. 2008, 84, 548–558. [Google Scholar] [CrossRef] [PubMed]
- Peters, C.; Brown, S. Antibody–drug conjugates as novel anti-cancer chemotherapeutics. Biosci. Rep. 2015, 35, e00225. [Google Scholar] [CrossRef] [Green Version]
- Williams, R.J.; Tse, T.; Dipiazza, K.; Zarin, D.A. Terminated Trials in the ClinicalTrials.gov Results Database: Evaluation of Availability of Primary Outcome Data and Reasons for Termination. PLoS ONE 2015, 10, e0127242. [Google Scholar] [CrossRef] [Green Version]
- Donaghy, H. Effects of antibody, drug and linker on the preclinical and clinical toxicities of antibody-drug conjugates. MAbs 2016, 8, 659–671. [Google Scholar] [CrossRef]
- Nejadmoghaddam, M.-R.; Minai-Tehrani, A.; Ghahremanzadeh, R.; Mahmoudi, M.; Dinarvand, R.; Zarnani, A.-H. Antibody-Drug Conjugates: Possibilities and Challenges. Avicenna J. Med. Biotechnol. 2019, 11, 3–23. [Google Scholar]
- Coats, S.; Williams, M.; Kebble, B.; Dixit, R.; Tseng, L.; Yao, N.-S.; Tice, D.A.; Soria, J.-C. Antibody–Drug Conjugates: Future Directions in Clinical and Translational Strategies to Improve the Therapeutic Index. Clin. Cancer Res. 2019, 25, 5441–5448. [Google Scholar] [CrossRef] [Green Version]
- Wolska-Washer, A.; Robak, T. Safety and Tolerability of Antibody-Drug Conjugates in Cancer. Drug Saf. 2019, 42, 295–314. [Google Scholar] [CrossRef] [Green Version]
- Masters, J.C.; Nickens, D.J.; Xuan, D.; Shazer, R.L.; Amantea, M. Clinical toxicity of antibody drug conjugates: A meta-analysis of payloads. Investig. New Drugs 2018, 36, 121–135. [Google Scholar] [CrossRef]
- Dokmanovic, M.; El Zarrad, K.; Hirsch, D.S.; Wu, W.J. Antibody-drug conjugates as therapeutic agents in oncology: Overview and perspectives. In Frontiers in Anti-Cancer Drug Discovery; Atta-Ur-Rahman, M., Choudhary, I., Eds.; Bentham Science Publishers: Oak Park, IL, USA, 2013; pp. 139–189. [Google Scholar]
- Lansita, J.A.; Burke, J.M.; Apgar, J.F.; Mounho-Zamora, B. An introduction to the regulatory and nonclinical aspects of the nonclinical development of antibody drug conjugates. Pharm. Res. 2015, 32, 3584–3592. [Google Scholar] [CrossRef] [PubMed]
- Saber, H.; Leighton, J.K. An FDA oncology analysis of antibody-drug conjugates. Regul. Toxicol. Pharmacol. 2015, 71, 444–452. [Google Scholar] [CrossRef]
- Casi, G.; Neri, D. Antibody–Drug Conjugates and Small Molecule–Drug Conjugates: Opportunities and Challenges for the Development of Selective Anticancer Cytotoxic Agents. J. Med. Chem. 2015, 58, 8751–8761. [Google Scholar] [CrossRef]
- Baker, M.P.; Reynolds, H.M.; Lumicisi, B.; Bryson, C.J. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self Nonself 2010, 1, 314–322. [Google Scholar] [CrossRef] [Green Version]
- Guffroy, M.; Falahatpisheh, H.; Finkelstein, M. Improving the Safety Profile of ADCs. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin, M., Ed.; Springer International Publishing: Cham, Germany, 2018; pp. 45–71. [Google Scholar]
- Palanca-Wessels, M.C.A.; Czuczman, M.; Salles, G.; Assouline, S.; Sehn, L.H.; Flinn, I.; Patel, M.R.; Sangha, R.; Hagenbeek, A.; Advani, R.; et al. Safety and activity of the anti-CD79B antibody–drug conjugate polatuzumab vedotin in relapsed or refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukaemia: A phase 1 study. Lancet Oncol. 2015, 16, 704–715. [Google Scholar] [CrossRef]
- Rosenberg, J.; Sridhar, S.S.; Zhang, J.; Smith, D.; Ruether, D.; Flaig, T.W.; Baranda, J.; Lang, J.; Plimack, E.R.; Sangha, R.; et al. EV-101: A Phase I Study of Single-Agent Enfortumab Vedotin in Patients with Nectin-4–Positive Solid Tumors, Including Metastatic Urothelial Carcinoma. J. Clin. Oncol. 2020, 38, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; Concin, N.; Hong, D.S.; Thistlethwaite, F.C.; Machiels, J.-P.; Arkenau, H.-T.; Plummer, R.; Jones, R.H.; Nielsen, D.; Windfeld, K.; et al. Tisotumab vedotin in patients with advanced or metastatic solid tumours (InnovaTV 201): A first-in-human, multicentre, phase 1–2 trial. Lancet Oncol. 2019, 20, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Saber, H.; Simpson, N.; Ricks, T.K.; Leighton, J.K. An FDA oncology analysis of toxicities associated with PBD-containing antibody-drug conjugates. Regul. Toxicol. Pharm. 2019, 107, 104429. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Liu, K.; Wang, K.; Zhu, H. Treatment-related adverse events of antibody–drug conjugates in clinical trials: A systematic review and meta-analysis. Cancer 2023, 129, 283–295. [Google Scholar] [CrossRef]
- Beck, A.; Goetsch, L.; Dumontet, C.; Corvaia, N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat. Rev. Drug Discov. 2017, 16, 315–337. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Jiang, F.; Lu, A.; Zhang, G. Linkers Having a Crucial Role in Antibody–Drug Conjugates. Int. J. Mol. Sci. 2016, 17, 561. [Google Scholar] [CrossRef]
- Bargh, J.D.; Isidro-Llobet, A.; Parker, J.S.; Spring, D.R. Cleavable linkers in antibody–drug conjugates. Chem. Soc. Rev. 2019, 48, 4361–4374. [Google Scholar] [CrossRef]
- Tumey, L.N.; Rago, B.; Han, X. In vivo biotransformations of antibody-drug conjugates. Bioanalysis 2015, 7, 1649–1664. [Google Scholar] [CrossRef]
- Drake, P.M.; Rabuka, D. Recent Developments in ADC Technology: Preclinical Studies Signal Future Clinical Trends. BioDrugs 2017, 31, 521–531. [Google Scholar] [CrossRef] [Green Version]
- Dorywalska, M.; Dushin, R.; Moine, L.; Farias, S.E.; Zhou, D.; Navaratnam, T.; Lui, V.; Hasa-Moreno, A.; Casas, M.G.; Tran, T.-T.; et al. Molecular Basis of Valine-Citrulline-PABC Linker Instability in Site-Specific ADCs and Its Mitigation by Linker Design. Mol. Cancer Ther. 2016, 15, 958–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillow, T.H.; Sadowsky, J.D.; Zhang, D.; Yu, S.-F.; Del Rosario, G.; Xu, K.; He, J.; Bhakta, S.; Ohri, R.; Kozak, K.R.; et al. Decoupling stability and release in disulfide bonds with antibody-small molecule conjugates. Chem. Sci. 2017, 8, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCombs, J.R.; Owen, S.C. Antibody Drug Conjugates: Design and Selection of Linker, Payload and Conjugation Chemistry. AAPS J. 2015, 17, 339–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mahalingaiah, P.K.; Ciurlionis, R.; Durbin, K.R.; Yeager, R.L.; Philip, B.K.; Bawa, B.; Mantena, S.R.; Enright, B.P.; Liguori, M.J.; van Vleet, T.R.; et al. Potential mechanisms of target-independent uptake and toxicity of antibody-drug conjugates. Pharmacol. Ther. 2019, 200, 110–125. [Google Scholar] [CrossRef]
- Singh, A.P.; Sharma, S.; Shah, D.K. Quantitative characterization of in vitro bystander effect of antibody-drug conjugates. J. Pharmacokinet. Pharmacodyn. 2016, 43, 567–582. [Google Scholar] [CrossRef] [Green Version]
- Polson, A.G.; Calemine-Fenaux, J.; Chan, P.; Chang, W.; Christensen, E.; Clark, S.; de Sauvage, F.J.; Eaton, D.; Elkins, K.; Elliott, J.M.; et al. Antibody-drug conjugates for the treatment of non-Hodgkin’s lymphoma: Target and linker-drug selection. Cancer Res. 2009, 69, 2358–2364. [Google Scholar] [CrossRef] [Green Version]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F.; et al. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Ponte, J.F.; Yoder, N.C.; Laleau, R.; Coccia, J.; Lanieri, L.; Qiu, Q.; Wu, R.; Hong, E.; Bogalhas, M.; et al. Effects of Drug–Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody–Maytansinoid Conjugates. Bioconjugate Chem. 2017, 28, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Boswell, C.A.; Tesar, D.B.; Mukhyala, K.; Theil, F.-P.; Fielder, P.J.; Khawli, L.A. Effects of Charge on Antibody Tissue Distribution and Pharmacokinetics. Bioconjugate Chem. 2010, 21, 2153–2163. [Google Scholar] [CrossRef] [PubMed]
- Stüber, J.C.; Rechberger, K.F.; Miladinović, S.M.; Pöschinger, T.; Zimmermann, T.; Villenave, R.; Eigenmann, M.J.; Kraft, T.E.; Shah, D.K.; Kettenberger, H.; et al. Impact of charge patches on tumor disposition and biodistribution of therapeutic antibodies. AAPS Open 2022, 8, 3. [Google Scholar] [CrossRef]
- Liu, S.; Verma, A.; Kettenberger, H.; Richter, W.F.; Shah, D.K. Effect of variable domain charge on in vitro and in vivo disposition of monoclonal antibodies. mAbs 2021, 13, 1993769. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Atkinson, J.; Gulesserian, S.; Zeng, Z.; Nater, J.; Ou, J.; Yang, P.; Morrison, K.; Coleman, J.; Malik, F.; et al. Modulation of Macropinocytosis-Mediated Internalization Decreases Ocular Toxicity of Antibody–Drug Conjugates. Cancer Res. 2018, 78, 2115–2126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapur, R.; Einarsdottir, H.K.; Vidarsson, G. IgG-effector functions: “The Good, The Bad and The Ugly”. Immunol. Lett. 2014, 160, 139–144. [Google Scholar] [CrossRef]
- Uppal, H.; Doudement, E.; Mahapatra, K.; Darbonne, W.C.; Bumbaca, D.; Shen, B.-Q.; Du, X.; Saad, O.; Bowles, K.; Olsen, S.; et al. Potential Mechanisms for Thrombocytopenia Development with Trastuzumab Emtansine (T-DM1). Clin. Cancer Res. 2015, 21, 123–133. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Gulesserian, S.; Ganesan, S.K.; Ou, J.; Morrison, K.; Zeng, Z.; Robles, V.; Snyder, J.; Do, L.; Aviña, H.; et al. Inhibition of Megakaryocyte Differentiation by Antibody–Drug Conjugates (ADCs) is Mediated by Macropinocytosis: Implications for ADC-induced Thrombocytopenia. Mol. Cancer Ther. 2017, 16, 1877–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoyama, M.; Tada, M.; Yokoo, H.; Demizu, Y.; Ishii-Watabe, A. Fcγ Receptor-Dependent Internalization and Off-Target Cytotoxicity of Antibody-Drug Conjugate Aggregates. Pharm. Res. 2022, 39, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Tada, M.; Aoyama, M.; Ishii-Watabe, A. Fcγ Receptor Activation by Human Monoclonal Antibody Aggregates. J. Pharm. Sci. 2020, 109, 576–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmadi, M.; Bryson, C.J.; Cloake, E.A.; Welch, K.; Filipe, V.; Romeijn, S.; Hawe, A.; Jiskoot, W.; Baker, M.P.; Fogg, M.H. Small Amounts of Sub-Visible Aggregates Enhance the Immunogenic Potential of Monoclonal Antibody Therapeutics. Pharm. Res. 2015, 32, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Ohri, R.; Bhakta, S.; Fourie-O’Donohue, A.; dela Cruz-Chuh, J.; Tsai, S.P.; Cook, R.; Wei, B.; Ng, C.; Wong, A.W.; Bos, A.B.; et al. High-Throughput Cysteine Scanning To Identify Stable Antibody Conjugation Sites for Maleimide- and Disulfide-Based Linkers. Bioconjugate Chem. 2018, 29, 473–485. [Google Scholar] [CrossRef]
- Buecheler, J.W.; Winzer, M.; Tonillo, J.; Weber, C.; Gieseler, H. Impact of Payload Hydrophobicity on the Stability of Antibody–Drug Conjugates. Mol. Pharm. 2018, 15, 2656–2664. [Google Scholar] [CrossRef]
- Mills, B.J.; Kruger, T.; Bruncko, M.; Zhang, X.; Jameel, F. Effect of Linker-Drug Properties and Conjugation Site on the Physical Stability of ADCs. J. Pharm. Sci. 2020, 109, 1662–1672. [Google Scholar] [CrossRef]
- Bruggeman, C.W.; Houtzager, J.; Dierdorp, B.; Kers, J.; Pals, S.T.; Lutter, R.; van Gulik, T.; den Haan, J.M.M.; van den Berg, T.K.; van Bruggen, R.; et al. Tissue-specific expression of IgG receptors by human macrophages ex vivo. PLoS ONE 2019, 14, e0223264. [Google Scholar] [CrossRef]
- Hackshaw, M.D.; Danysh, H.E.; Singh, J.; Ritchey, M.E.; Ladner, A.; Taitt, C.; Camidge, D.R.; Iwata, H.; Powell, C.A. Incidence of pneumonitis/interstitial lung disease induced by HER2-targeting therapy for HER2-positive metastatic breast cancer. Breast Cancer Res. Treat. 2020, 183, 23–39. [Google Scholar] [CrossRef]
- Kumagai, K.; Aida, T.; Tsuchiya, Y.; Kishino, Y.; Kai, K.; Mori, K. Interstitial pneumonitis related to trastuzumab deruxtecan, a human epidermal growth factor receptor 2-targeting Ab-drug conjugate, in monkeys. Cancer Sci. 2020, 111, 4636–4645. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.; Norvell, T.M.; Tosi, M.F.; Emancipator, S.N.; Konstan, M.W.; Schreiber, J.R. Tissue-specific Fc gamma and complement receptor expression by alveolar macrophages determines relative importance of IgG and complement in promoting phagocytosis of Pseudomonas aeruginosa. Pediatr. Res. 1994, 35, 68–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conte, P.; Ascierto, P.A.; Patelli, G.; Danesi, R.; Vanzulli, A.; Sandomenico, F.; Tarsia, P.; Cattelan, A.; Comes, A.; De Laurentiis, M.; et al. Drug-induced interstitial lung disease during cancer therapies: Expert opinion on diagnosis and treatment. ESMO Open 2022, 7, 100404. [Google Scholar] [CrossRef] [PubMed]
- Gorovits, B.; Krinos-Fiorotti, C. Proposed mechanism of off-target toxicity for antibody–drug conjugates driven by mannose receptor uptake. Cancer Immunol. Immunother. 2013, 62, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Goetze, A.M.; Liu, Y.D.; Zhang, Z.; Shah, B.; Lee, E.; Bondarenko, P.V.; Flynn, G.C. High-mannose glycans on the Fc region of therapeutic IgG antibodies increase serum clearance in humans. Glycobiology 2011, 21, 949–959. [Google Scholar] [CrossRef] [Green Version]
- Kogelberg, H.; Tolner, B.; Sharma, S.K.; Lowdell, M.W.; Qureshi, U.; Robson, M.; Hillyer, T.; Pedley, R.B.; Vervecken, W.; Contreras, R.; et al. Clearance mechanism of a mannosylated antibody-enzyme fusion protein used in experimental cancer therapy. Glycobiology 2007, 17, 36–45. [Google Scholar] [CrossRef] [Green Version]
- Guffroy, M.; Falahatpisheh, H.; Biddle, K.; Kreeger, J.; Obert, L.; Walters, K.; Goldstein, R.; Boucher, G.; Coskran, T.; Reagan, W.; et al. Liver Microvascular Injury and Thrombocytopenia of Antibody–Calicheamicin Conjugates in Cynomolgus Monkeys—Mechanism and Monitoring. Clin. Cancer Res. 2017, 23, 1760–1770. [Google Scholar] [CrossRef] [Green Version]
- Drago, J.Z.; Modi, S.; Chandarlapaty, S. Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 2021, 18, 327–344. [Google Scholar] [CrossRef]
- S.A.A.P.U. Padcev Inc. (Enfortumab Vedotinejfv) for Injection [Prescribing Information]. Available online: https://astellas.us/docs/PADCEV_label.pdf (accessed on 2 April 2022).
- Challita-Eid, P.M.; Satpayev, D.; Yang, P.; An, Z.; Morrison, K.; Shostak, Y.; Raitano, A.; Nadell, R.; Liu, W.; Lortie, D.R.; et al. Enfortumab Vedotin Antibody–Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res. 2016, 76, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- FDA: US Prescribing Information: Brentuximab Vedotin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/125388s100lbl.pdf (accessed on 2 April 2022).
- FDA: US Prescribing Information: Polatuzumab Vedotin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761121s000lbl.pdf (accessed on 2 April 2022).
- TIVDAK (Tisotumab Vedoin-tftv): US Prescribing Information. Available online: https://seagendocs.com/Tivdak_Full_Ltr_Master.pdf (accessed on 2 April 2022).
- Modi, S.; Saura, C.; Yamashita, T.; Park, Y.H.; Kim, S.-B.; Tamura, K.; Andre, F.; Iwata, H.; Ito, Y.; Tsurutani, J.; et al. Trastuzumab Deruxtecan in Previously Treated HER2-Positive Breast Cancer. N. Engl. J. Med. 2020, 382, 610–621. [Google Scholar] [CrossRef]
- FDA: US Prescribing Information: Trastuzumab Emtansine. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/125427lbl.pdf (accessed on 2 April 2022).
- Banerji, U.; van Herpen, C.M.L.; Saura, C.; Thistlethwaite, F.; Lord, S.; Moreno, V.; Macpherson, I.R.; Boni, V.; Rolfo, C.; De Vries, E.G.E.; et al. Trastuzumab duocarmazine in locally advanced and metastatic solid tumours and HER2-expressing breast cancer: A phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019, 20, 1124–1135. [Google Scholar] [CrossRef] [Green Version]
- Siegel, P.; Rose, A.; Maric, M.G.; Siegel, P.M. Glycoprotein non-metastatic b (GPNMB): A metastatic mediator and emerging therapeutic target in cancer. OncoTargets Ther. 2013, 2013, 839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ott, P.A.; Hamid, O.; Pavlick, A.C.; Kluger, H.; Kim, K.B.; Boasberg, P.D.; Simantov, R.; Crowley, E.; Green, J.A.; Hawthorne, T.; et al. Phase I/II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Patients with Advanced Melanoma. J. Clin. Oncol. 2014, 32, 3659–3666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardley, D.A.; Weaver, R.; Melisko, M.E.; Saleh, M.N.; Arena, F.P.; Forero, A.; Cigler, T.; Stopeck, A.; Citrin, D.; Oliff, I.; et al. EMERGE: A Randomized Phase II Study of the Antibody-Drug Conjugate Glembatumumab Vedotin in Advanced Glycoprotein NMB–Expressing Breast Cancer. J. Clin. Oncol. 2015, 33, 1609–1619. [Google Scholar] [CrossRef]
- L’Italien, L.; Orozco, O.; Abrams, T.; Cantagallo, L.; Connor, A.; Desai, J.; Ebersbach, H.; Gelderblom, H.; Hoffmaster, K.; Lees, E.; et al. Mechanistic Insights of an Immunological Adverse Event Induced by an Anti-KIT Antibody Drug Conjugate and Mitigation Strategies. Clin. Cancer Res. 2018, 24, 3465–3474. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.F.; Moore, K.N.; Birrer, M.J.; Berlin, S.; Matulonis, U.A.; Infante, J.R.; Wolpin, B.; Poon, K.A.; Firestein, R.; Xu, J.; et al. Phase I study of safety and pharmacokinetics of the anti-MUC16 antibody–drug conjugate DMUC5754A in patients with platinum-resistant ovarian cancer or unresectable pancreatic cancer. Ann. Oncol. 2016, 27, 2124–2130. [Google Scholar] [CrossRef]
- Argu’Eso, P.; Spurr-Michaud, S.; Russo, C.L.; Tisdale, A.; Gipson, I.K. MUC16 Mucin Is Expressed by the Human Ocular Surface Epithelia and Carries the H185 Carbohydrate Epitope. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2487–2495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stepan, L.P.; Trueblood, E.S.; Hale, K.; Babcook, J.; Borges, L.; Sutherland, C.L. Expression of Trop2 Cell Surface Glycoprotein in Normal and Tumor Tissues. J. Histochem. Cytochem. 2011, 59, 701–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients with Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef]
- Fenn, K.M.; Kalinsky, K. Sacituzumab govitecan: Antibody-drug conjugate in triple-negative breast cancer and other solid tumors. Drugs Today 2019, 55, 575–585. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Beyer, C.F.; Lindh, D.; Upeslacis, J.; Flowers, D.A.; Bernstein, I. An anti-CD33 antibody-calicheamicin conjugate for treatment of acute myeloid leukemia. Choice of linker. Bioconjugate Chem. 2002, 13, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Hamann, P.R.; Hinman, L.M.; Hollander, I.; Beyer, C.F.; Lindh, D.; Holcomb, R.; Hallett, W.; Tsou, H.-R.; Upeslacis, J.; Shochat, D.; et al. Gemtuzumab Ozogamicin, A Potent and Selective Anti-CD33 Antibody−Calicheamicin Conjugate for Treatment of Acute Myeloid Leukemia. Bioconjugate Chem. 2002, 13, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L.; Appelbaum, F.R.; Spielberger, R.T.; Forman, S.J.; Flowers, D.; Smith, F.O.; Shannon-Dorcy, K.; Berger, M.S.; Bernstein, I.D. Selective Ablation of Acute Myeloid Leukemia Using Antibody-Targeted Chemotherapy: A Phase I Study of an Anti-CD33 Calicheamicin Immunoconjugate. Blood 1999, 93, 3678–3684. [Google Scholar] [CrossRef] [PubMed]
- Sievers, E.L. Efficacy and safety of gemtuzumab ozogamicin in patients with CD33-positive acute myeloid leukaemia in first relapse. Expert Opin. Biol. Ther. 2001, 1, 893–901. [Google Scholar] [CrossRef]
- Rowe, J.M.; Lowenberg, B. Gemtuzumab ozogamicin in acute myeloid leukemia: A remarkable saga about an active drug. Blood 2013, 121, 4838–4841. [Google Scholar] [CrossRef]
- Petersdorf, S.H.; Kopecky, K.J.; Slovak, M.; Willman, C.; Nevill, T.; Brandwein, J.; Larson, R.A.; Erba, H.P.; Stiff, P.J.; Stuart, R.K.; et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood 2013, 121, 4854–4860. [Google Scholar] [CrossRef] [Green Version]
- Amadori, S.; Suciu, S.; Selleslag, D.; Aversa, F.; Gaidano, G.; Musso, M.; Annino, L.; Venditti, A.; Voso, M.T.; Mazzone, C.; et al. Gemtuzumab Ozogamicin Versus Best Supportive Care in Older Patients with Newly Diagnosed Acute Myeloid Leukemia Unsuitable for Intensive Chemotherapy: Results of the Randomized Phase III Eortc-Gimema Aml-19 Trial. J. Clin. Oncol. 2016, 34, 972–979. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.; Pautas, C.; Terré, C.; Raffoux, E.; Turlure, P.; Caillot, D.; Legrand, O.; Thomas, X.; Gardin, C.; Gogat-Marchant, K.; et al. Gemtuzumab ozogamicin for de novo acute myeloid leukemia: Final efficacy and safety updates from the open-label, phase III ALFA-0701 trial. Haematologica 2019, 104, 113–119. [Google Scholar] [CrossRef] [Green Version]
- Norsworthy, K.J.; Ko, C.-W.; Lee, J.E.; Liu, J.; John, C.S.; Przepiorka, D.; Farrell, A.T.; Pazdur, R. FDA Approval Summary: Mylotarg for Treatment of Patients with Relapsed or Refractory CD33-Positive Acute Myeloid Leukemia. Oncolgist 2018, 23, 1103–1108. [Google Scholar] [CrossRef] [Green Version]
- FDA: US Prescribing Information: Gemtuzumab Ozogamicin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761060lbl.pdf (accessed on 2 April 2022).
- Larson, R.A.; Sievers, E.L.; Stadtmauer, E.A.; Löwenberg, B.; Estey, E.H.; Dombret, H.; Theobald, M.; Voliotis, D.; Bennett, J.M.; Richie, M.; et al. Final report of the efficacy and safety of gemtuzumab ozogamicin (Mylotarg) in patients with CD33-positive acute myeloid leukemia in first recurrence. Cancer 2005, 104, 1442–1452. [Google Scholar] [CrossRef] [PubMed]
- Fenton, C.; Perry, C.M. Gemtuzumab Ozogamicin. Drugs 2005, 65, 2405–2427. [Google Scholar] [CrossRef]
- Maniecki, M.B.; Hasle, H.; Bendix, K.; Møller, H.J. Is hepatotoxicity in patients treated with gemtuzumabozogamicin due to specific targeting of hepatocytes? Leuk. Res. 2011, 35, e84–e86. [Google Scholar] [CrossRef]
- Rajvanshi, P.; Shulman, H.M.; Sievers, E.L.; McDonald, G.B. Hepatic sinusoidal obstruction after gemtuzumab ozogamicin (Mylotarg) therapy. Blood 2002, 99, 2310–2314. [Google Scholar] [CrossRef] [PubMed]
- Godwin, C.D.; McDonald, G.B.; Walter, R.B. Sinusoidal obstruction syndrome following CD33-targeted therapy in acute myeloid leukemia. Blood 2017, 129, 2330–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, M.; Hecht, K.; Reif, S.; Pelka-Fleischer, R.; Pfister, K.; Schmetzer, H. Expression and prognostic value of hemopoietic cytokine receptors in acute myeloid leukemia (AML): Implications for future therapeutical strategies. Eur. J. Haematol. 2004, 72, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Van Rhenen, A.; van Dongen, G.A.M.S.; Kelder, A.; Rombouts, E.J.; Feller, N.; Moshaver, B.; Walsum, M.S.-V.; Zweegman, S.; Ossenkoppele, G.J.; Jan Schuurhuis, G. The novel AML stem cell–associated antigen CLL-1 aids in discrimination between normal and leukemic stem cells. Blood 2007, 110, 2659–2666. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shor, B.; Gerber, H.-P.; Sapra, P. Preclinical and clinical development of inotuzumab-ozogamicin in hematological malignancies. Mol. Immunol. 2015, 67, 107–116. [Google Scholar] [CrossRef]
- Deangelo, D.J.; Stock, W.; Stein, A.S.; Shustov, A.; Liedtke, M.; Schiffer, C.A.; Vandendries, E.; Liau, K.; Ananthakrishnan, R.; Boni, J.; et al. Inotuzumab ozogamicin in adults with relapsed or refractory CD22-positive acute lymphoblastic leukemia: A phase 1/2 study. Blood Adv. 2017, 1, 1167–1180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kantarjian, H.M.; Deangelo, D.J.; Stelljes, M.; Martinelli, G.; Liedtke, M.; Stock, W.; Gökbuget, N.; O’Brien, S.; Wang, K.; Wang, T.; et al. Inotuzumab Ozogamicin versus Standard Therapy for Acute Lymphoblastic Leukemia. N. Engl. J. Med. 2016, 375, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Kantarjian, H.M.; de Angelo, D.J.; Advani, A.S.; Stelljes, M.; Kebriaei, P.; Cassaday, R.D.; Merchant, A.A.; Fujishima, N.; Uchida, T.; Calbacho, M.; et al. Hepatic adverse event profile of inotuzumab ozogamicin in adult patients with relapsed or refractory acute lymphoblastic leukaemia: Results from the open-label, randomised, phase 3 INO-VATE study. Lancet Haematol. 2017, 4, e387–e398. [Google Scholar] [CrossRef]
- FDA: US Prescribing Information: Inotuzumab Ozogamicin. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/761040s000lbl.pdf (accessed on 2 April 2022).
- Ansell, S.M. Brentuximab vedotin. Blood 2014, 124, 3197–3200. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab Vedotin (SGN-35) for Relapsed CD30-Positive Lymphomas. N. Engl. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Younes, A.; Gopal, A.K.; Smith, S.E.; Ansell, S.M.; Rosenblatt, J.D.; Savage, K.J.; Ramchandren, R.; Bartlett, N.L.; Cheson, B.D.; de Vos, S.; et al. Results of a Pivotal Phase II Study of Brentuximab Vedotin for Patients with Relapsed or Refractory Hodgkin’s Lymphoma. J. Clin. Oncol. 2012, 30, 2183–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pro, B.; Advani, R.; Brice, P.; Bartlett, N.L.; Rosenblatt, J.D.; Illidge, T.; Matous, J.; Ramchandren, R.; Fanale, M.; Connors, J.M.; et al. Brentuximab vedotin (SGN-35) in patients with relapsed or refractory systemic anaplastic large-cell lymphoma: Results of a phase II study. J. Clin. Oncol. 2012, 30, 2190–2196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moskowitz, C.H.; Nademanee, A.; Masszi, T.; Agura, E.; Holowiecki, J.; Abidi, M.H.; Chen, A.I.; Stiff, P.; Gianni, A.M.; Carella, A.; et al. Brentuximab vedotin as consolidation therapy after autologous stem-cell transplantation in patients with Hodgkin’s lymphoma at risk of relapse or progression (AETHERA): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2015, 385, 1853–1862. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Polatuzumab Vedotin: First Global Approval. Drugs 2019, 79, 1467–1475. [Google Scholar] [CrossRef] [Green Version]
- Morschhauser, F.; Flinn, I.W.; Advani, R.; Sehn, L.H.; Diefenbach, C.; Kolibaba, K.; Press, O.W.; Salles, G.; Tilly, H.; Chen, A.I.; et al. Polatuzumab vedotin or pinatuzumab vedotin plus rituximab in patients with relapsed or refractory non-Hodgkin lymphoma: Final results from a phase 2 randomised study (ROMULUS). Lancet Haematol. 2019, 6, e254–e265. [Google Scholar] [CrossRef]
- Sehn, L.H.; Herrera, A.F.; Flowers, C.R.; Kamdar, M.K.; McMillan, A.; Hertzberg, M.; Assouline, S.; Kim, T.M.; Kim, W.S.; Ozcan, M.; et al. Polatuzumab Vedotin in Relapsed or Refractory Diffuse Large B-Cell Lymphoma. J. Clin. Oncol. 2020, 38, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Terui, Y.; Rai, S.; Izutsu, K.; Yamaguchi, M.; Takizawa, J.; Kuroda, J.; Ishikawa, T.; Kato, K.; Suehiro, Y.; Fukuhara, N.; et al. A phase 2 study of polatuzumab vedotin + bendamustine + rituximab in relapsed/refractory diffuse large B-cell lymphoma. Cancer Sci. 2021, 112, 2845–2854. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; O’Donnell, P.H.; Balar, A.V.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.M.; et al. Pivotal Trial of Enfortumab Vedotin in Urothelial Carcinoma After Platinum and Anti-Programmed Death 1/Programmed Death Ligand 1 Therapy. J. Clin. Oncol. 2019, 37, 2592–2600. [Google Scholar] [CrossRef]
- Powles, T.; Rosenberg, J.E.; Sonpavde, G.P.; Loriot, Y.; Durán, I.; Lee, J.-L.; Matsubara, N.; Vulsteke, C.; Castellano, D.; Wu, C.; et al. Enfortumab Vedotin in Previously Treated Advanced Urothelial Carcinoma. N. Engl. J. Med. 2021, 384, 1125–1135. [Google Scholar] [CrossRef]
- Lacouture, M.E.; Patel, A.B.; Rosenberg, J.E.; O’Donnell, P.H. Management of Dermatologic Events Associated with the Nectin-4-directed Antibody-Drug Conjugate Enfortumab Vedotin. Oncolgist 2022, 27, e223–e232. [Google Scholar] [CrossRef]
- Ott, P.A.; Pavlick, A.C.; Johnson, D.B.; Hart, L.L.; Infante, J.R.; Luke, J.J.; Lutzky, J.; Rothschild, N.E.; Spitler, L.E.; Cowey, C.L.; et al. A phase 2 study of glembatumumab vedotin, an antibody-drug conjugate targeting glycoprotein NMB, in patients with advanced melanoma. Cancer 2019, 125, 1113–1123. [Google Scholar] [CrossRef] [PubMed]
- Chang, E.; Weinstock, C.; Zhang, L.; Charlab, R.; Dorff, S.E.; Gong, Y.; Hsu, V.; Li, F.; Ricks, T.K.; Song, P.; et al. FDA Approval Summary: Enfortumab Vedotin for Locally Advanced or Metastatic Urothelial Carcinoma. Clin. Cancer Res. 2021, 27, 922–927. [Google Scholar] [CrossRef]
- Markham, A. Tisotumab Vedotin: First Approval. Drugs 2021, 81, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.L.; Lorusso, D.; Gennigens, C.; González-Martín, A.; Randall, L.; Cibula, D.; Lund, B.; Woelber, L.; Pignata, S.; Forget, F.; et al. Efficacy and safety of tisotumab vedotin in previously treated recurrent or metastatic cervical cancer (innovaTV 204/GOG-3023/ENGOT-cx6): A multicentre, open-label, single-arm, phase 2 study. Lancet Oncol. 2021, 22, 609–619. [Google Scholar] [CrossRef]
- Cho, Y.; Cao, X.; Shen, D.; Tuo, J.; Parver, L.M.; Rickles, F.R.; Chan, C.-C. Evidence for enhanced tissue factor expression in age-related macular degeneration. Lab. Investig. 2011, 91, 519–526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ando, R.; Kase, S.; Ohashi, T.; Dong, Z.; Fukuhara, J.; Kanda, A.; Murata, M.; Noda, K.; Kitaichi, N.; Ishida, S. Tissue factor expression in human pterygium. Mol. Vis. 2011, 17, 63–69. [Google Scholar]
- Trudel, S.; Lendvai, N.; Popat, R.; Voorhees, P.M.; Reeves, B.; Libby, E.N.; Richardson, P.G.; Anderson, L.D.; Sutherland, H.J.; Yong, K.; et al. Targeting B-cell maturation antigen with GSK2857916 antibody–drug conjugate in relapsed or refractory multiple myeloma (BMA117159): A dose escalation and expansion phase 1 trial. Lancet Oncol. 2018, 19, 1641–1653. [Google Scholar] [CrossRef]
- Lonial, S.; Lee, H.C.; Badros, A.; Trudel, S.; Nooka, A.K.; Chari, A.; Abdallah, A.-O.; Callander, N.; Lendvai, N.; Sborov, D.; et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM-2): A two-arm, randomised, open-label, phase 2 study. Lancet Oncol. 2020, 21, 207–221. [Google Scholar] [CrossRef]
- GlaxoSmithKline. BLENREP (Belantamab Mafodotin-Blmf): US Prescribing Information. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/761158s000lbl.pdf (accessed on 2 April 2022).
- GlaxoSmithKline. GSK Provides an Update on Blenrep (Belantamab Mafodotin-Blmf) US Marketing Authorisation. 2022. Available online: https://www.gsk.com/en-gb/media/press-releases/gsk-provides-update-on-blenrep-us-marketing-authorisation/. (accessed on 17 January 2023).
- Ballantyne, A.; Dhillon, S. Trastuzumab Emtansine: First Global Approval. Drugs 2013, 73, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Beeram, M.; Krop, I.E.; Burris, H.A.; Girish, S.R.; Yu, W.; Lu, M.W.; Holden, S.N.; Modi, S. A phase 1 study of weekly dosing of trastuzumab emtansine (T-DM1) in patients with advanced human epidermal growth factor 2–positive breast cancer. Cancer 2012, 118, 5733–5740. [Google Scholar] [CrossRef] [PubMed]
- Krop, I.E.; Beeram, M.; Modi, S.; Jones, S.F.; Holden, S.N.; Yu, W.; Girish, S.; Tibbitts, J.; Yi, J.-H.; Sliwkowski, M.X.; et al. Phase I Study of Trastuzumab-DM1, an HER2 Antibody-Drug Conjugate, Given Every 3 Weeks to Patients With HER2-Positive Metastatic Breast Cancer. J. Clin. Oncol. 2010, 28, 2698–2704. [Google Scholar] [CrossRef]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E.; et al. Trastuzumab Emtansine for HER2-Positive Advanced Breast Cancer. N. Engl. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.-H.; Guo, W.-Z.; Jin, Y.; Zhang, H.-P.; Pang, C.; Li, J.; Line, P.-D.; Zhang, S.-J. Recognition of HER2 expression in hepatocellular carcinoma and its significance in postoperative tumor recurrence. Cancer Med. 2019, 8, 1269–1278. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Endo, Y.; Shen, Y.; Rotstein, D.; Dokmanovic, M.; Mohan, N.; Mukhopadhyay, P.; Gao, B.; Pacher, P.; Wu, W.J. Ado-Trastuzumab Emtansine Targets Hepatocytes Via Human Epidermal Growth Factor Receptor 2 to Induce Hepatotoxicity. Mol. Cancer Ther. 2016, 15, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Endo, Y.; Takeda, K.; Mohan, N.; Shen, Y.; Jiang, J.; Rotstein, D.; Wu, W.J. Payload of T-DM1 binds to cell surface cytoskeleton-associated protein 5 to mediate cytotoxicity of hepatocytes. Oncotarget 2018, 9, 37200–37215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Endo, Y.; Mohan, N.; Dokmanovic, M.; Wu, W.J. Mechanisms contributing to ado-trastuzumab emtansine-induced toxicities: A gateway to better understanding of ADC-associated toxicities. Antib. Ther. 2021, 4, 55–59. [Google Scholar] [CrossRef]
- Moilanen, T.; Jokimäki, A.; Tenhunen, O.; Koivunen, J.P. Trastuzumab-induced cardiotoxicity and its risk factors in real-world setting of breast cancer patients. J. Cancer Res. Clin. Oncol. 2018, 144, 1613–1621. [Google Scholar] [CrossRef]
- National Toxicology Program. NTP Monograph: Developmental Effects and Pregnancy Outcomes Associated with Cancer Chemotherapy Use During Pregnancy. NTP Monogr. 2013, 2, i-214. [Google Scholar]
- Xia, L.-Y.; Hu, Q.-L.; Zhou, Q. Use of trastuzumab in treating breast cancer during pregnancy: A systematic review and meta-analysis. BMC Women’s Health 2021, 21, 169. [Google Scholar] [CrossRef] [PubMed]
- Ab, O.; Whiteman, K.R.; Bartle, L.M.; Sun, X.; Singh, R.; Tavares, D.; LaBelle, A.; Payne, G.; Lutz, R.J.; Pinkas, J.; et al. IMGN853, a Folate Receptor-alpha (FRalpha)-Targeting Antibody-Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRalpha-Expressing Tumors. Mol. Cancer Ther. 2015, 14, 1605–1613. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Borghaei, H.; O’Malley, D.M.; Jeong, W.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Matulonis, U.A.; Running, K.L.; Zhang, X.; et al. Phase 1 dose-escalation study of mirvetuximab soravtansine (IMGN853), a folate receptor α-targeting antibody-drug conjugate, in patients with solid tumors. Cancer 2017, 123, 3080–3087. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Oza, A.M.; Colombo, N.; Oaknin, A.; Scambia, G.; Lorusso, D.; Konecny, G.E.; Banerjee, S.; Murphy, C.G.; Tanyi, J.L.; et al. Phase III, randomized trial of mirvetuximab soravtansine versus chemotherapy in patients with platinum-resistant ovarian cancer: Primary analysis of Forward I. Ann. Oncol. 2021, 32, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Narayan, P.; Osgood, C.L.; Singh, H.; Chiu, H.-J.; Ricks, T.K.; Chiu Yuen Chow, E.; Qiu, J.; Song, P.; Yu, J.; Namuswe, F.; et al. FDA Approval Summary: Fam-Trastuzumab Deruxtecan-Nxki for the Treatment of Unresectable or Metastatic HER2-Positive Breast Cancer. Clin. Cancer Res. 2021, 27, 4478–4485. [Google Scholar] [CrossRef] [PubMed]
- Doi, T.; Iwata, H.; Tsurutani, J.; Takahashi, S.; Park, H.; Redfern, C.H.; Shitara, K.; Shimizu, C.; Taniguchi, H.; Iwasa, T.; et al. Single agent activity of DS-8201a, a HER2-targeting antibody-drug conjugate, in heavily pretreated HER2 expressing solid tumors. J. Clin. Oncol. 2017, 35, 108. [Google Scholar] [CrossRef]
- FDA: US Prescribing Information: Trastuzumab Deruxtecan. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/761139s000lbl.pdf (accessed on 2 April 2022).
- Goldenberg, D.M.; Sharkey, R.M. Sacituzumab Govitecan, A Novel, Third-Generation, Antibody-drug Conjugate (ADC) for Cancer Therapy. Expert Opin. Biol. Ther. 2020, 20, 871–885. [Google Scholar] [CrossRef]
- Starodub, A.N.; Ocean, A.J.; Shah, M.A.; Guarino, M.J.; Picozzi, V.J.; Vahdat, L.T.; Thomas, S.S.; Govindan, S.V.; Maliakal, P.P.; Wegener, W.A.; et al. First-in-Human Trial of a Novel Anti-Trop-2 Antibody-SN-38 Conjugate, Sacituzumab Govitecan, for the Treatment of Diverse Metastatic Solid Tumors. Clin. Cancer Res. 2015, 21, 3870–3878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocean, A.J.; Starodub, A.N.; Bardia, A.; Vahdat, L.T.; Isakoff, S.J.; Guarino, M.; Messersmith, W.A.; Picozzi, V.J.; Mayer, I.A.; Wegener, W.A.; et al. Sacituzumab govitecan (IMMU-132), an anti-Trop-2-SN-38 antibody-drug conjugate for the treatment of diverse epithelial cancers: Safety and pharmacokinetics. Cancer 2017, 123, 3843–3854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA: US Prescribing Information: Sacituzumab Govitecan. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761115s009lbl.pdf (accessed on 2 April 2022).
- FDA: US Prescribing Information: Irinotecan. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020571s048lbl.pdf (accessed on 2 April 2022).
- Lee, A. Loncastuximab Tesirine: First Approval. Drugs 2021, 81, 1229–1233. [Google Scholar] [CrossRef]
- Zammarchi, F.; Corbett, S.; Adams, L.; Tyrer, P.C.; Kiakos, K.; Janghra, N.; Marafioti, T.; Britten, C.E.; Havenith, C.E.G.; Chivers, S.; et al. ADCT-402, a PBD dimer–containing antibody drug conjugate targeting CD19-expressing malignancies. Blood 2018, 131, 1094–1105. [Google Scholar] [CrossRef] [Green Version]
- Hamadani, M.; Radford, J.; Carlo-Stella, C.; Caimi, P.F.; Reid, E.; O’Connor, O.A.; Feingold, J.M.; Ardeshna, K.M.; Townsend, W.; Solh, M.; et al. Final results of a phase 1 study of loncastuximab tesirine in relapsed/refractory B-cell non-Hodgkin lymphoma. Blood 2021, 137, 2634–2645. [Google Scholar] [CrossRef]
- Caimi, P.F.; Ai, W.; Alderuccio, J.P.; Ardeshna, K.M.; Hamadani, M.; Hess, B.; Kahl, B.S.; Radford, J.; Solh, M.; Stathis, A.; et al. Loncastuximab tesirine in relapsed or refractory diffuse large B-cell lymphoma (LOTIS-2): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2021, 22, 790–800. [Google Scholar] [CrossRef] [PubMed]
- FDA: US Prescribing Information: Loncastuximab Tesirine. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/761196s000lbl.pdf (accessed on 2 April 2022).
- Elgersma, R.C.; Coumans, R.G.E.; Huijbregts, T.; Menge, W.M.P.B.; Joosten, J.A.F.; Spijker, H.J.; de Groot, F.M.H.; van der Lee, M.M.C.; Ubink, R.; van den Dobbelsteen, D.J.; et al. Design, Synthesis, and Evaluation of Linker-Duocarmycin Payloads: Toward Selection of HER2-Targeting Antibody–Drug Conjugate SYD985. Mol. Pharm. 2015, 12, 1813–1835. [Google Scholar] [CrossRef] [PubMed]
- Saura Manich, C.; O’Shaughnessy, J.; Aftimos, P.G.; van den Tweel, E.; Oesterholt, M.; Escrivá-de-Romaní, S.I.; Quenel Tueux, N.; Tan, T.J.; Lim, J.S.; Ladoire, S.; et al. LBA15 Primary outcome of the phase III SYD985.002/TULIP trial comparing [vic-]trastuzumab duocarmazine to physician’s choice treatment in patients with pre-treated HER2-positive locally advanced or metastatic breast cancer. Ann. Oncol. 2021, 32, S1288. [Google Scholar] [CrossRef]
- Jiang, J.; Li, S.; Shan, X.; Wang, L.; Ma, J.; Huang, M.; Dong, L.; Chen, F. Preclinical safety profile of disitamab vedotin: A novel anti-HER2 antibody conjugated with MMAE. Toxicol. Lett. 2020, 324, 30–37. [Google Scholar] [CrossRef]
- Deeks, E.D. Disitamab Vedotin: First Approval. Drugs 2021, 81, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Shen, L.; Wang, W.; Fang, J. Safety, pharmacokinetics and efficacy of RC48-ADC in a phase I study in patients with HER2-overexpression advanced solid cancer. J. Clin. Oncol. 2018, 36, e16059. [Google Scholar] [CrossRef]
- Actor, J.K. 2-Cells and Organs of the Immune System. In Elsevier’s Integrated Review Immunology and Microbiology, 2nd ed.; Actor, J.K., Ed.; W.B. Saunders: Philadelphia, PA, USA, 2012; pp. 7–16. [Google Scholar]
- Summers, C.; Rankin, S.M.; Condliffe, A.M.; Singh, N.; Peters, A.M.; Chilvers, E.R. Neutrophil kinetics in health and disease. Trends Immunol. 2010, 31, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Bekkering, S. Another look at the life of a neutrophil. World J. Hematol. 2013, 2, 44. [Google Scholar] [CrossRef]
- Lahoz-Beneytez, J.; Elemans, M.; Zhang, Y.; Ahmed, R.; Salam, A.; Block, M.; Niederalt, C.; Asquith, B.; Macallan, D. Human neutrophil kinetics: Modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood 2016, 127, 3431–3438. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Gulesserian, S.; Malinao, M.C.; Ganesan, S.K.; Song, J.; Chang, M.S.; Williams, M.M.; Zeng, Z.; Mattie, M.; Mendelsohn, B.A.; et al. A Potential Mechanism for ADC-Induced Neutropenia: Role of Neutrophils in Their Own Demise. Mol. Cancer Ther. 2017, 16, 1866. [Google Scholar] [CrossRef] [Green Version]
- Kung Sutherland, M.S.; Walter, R.B.; Jeffrey, S.C.; Burke, P.J.; Yu, C.; Kostner, H.; Stone, I.; Ryan, M.C.; Sussman, D.; Lyon, R.P.; et al. SGN-CD33A: A novel CD33-targeting antibody-drug conjugate using a pyrrolobenzodiazepine dimer is active in models of drug-resistant AML. Blood 2013, 122, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Sutherland, M.K.; Yu, C.; Walter, R.B.; Westendorf, L.; Valliere-Douglass, J.; Pan, L.; Cronkite, A.; Sussman, D.; Klussman, K.; et al. Characterization of SGN-CD123A, A Potent CD123-Directed Antibody–Drug Conjugate for Acute Myeloid Leukemia. Mol. Cancer Ther. 2018, 17, 554–564. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Yu, S.-F.; del Rosario, G.; Leong, S.R.; Lee, G.Y.; Vij, R.; Chiu, C.; Liang, W.-C.; Wu, Y.; Chalouni, C.; et al. An Anti–CLL-1 Antibody–Drug Conjugate for the Treatment of Acute Myeloid Leukemia. Clin. Cancer Res. 2019, 25, 1358–1368. [Google Scholar] [CrossRef] [Green Version]
- Haubner, S.; Perna, F.; Köhnke, T.; Schmidt, C.; Berman, S.; Augsberger, C.; Schnorfeil, F.M.; Krupka, C.; Lichtenegger, F.S.; Liu, X.; et al. Coexpression profile of leukemic stem cell markers for combinatorial targeted therapy in AML. Leukemia 2019, 33, 64–74. [Google Scholar] [CrossRef]
- Grisold, W.; Cavaletti, G.; Windebank, A.J. Peripheral neuropathies from chemotherapeutics and targeted agents: Diagnosis, treatment, and prevention. Neuro Oncol. 2012, 14, iv45–iv54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galsky, M.D.; Eisenberger, M.; Moore-Cooper, S.; Kelly, W.K.; Slovin, S.F.; Delacruz, A.; Lee, Y.; Webb, I.J.; Scher, H.I. Phase I Trial of the Prostate-Specific Membrane Antigen–Directed Immunoconjugate MLN2704 in Patients with Progressive Metastatic Castration-Resistant Prostate Cancer. J. Clin. Oncol. 2008, 26, 2147–2154. [Google Scholar] [CrossRef] [PubMed]
- Milowsky, M.I.; Galsky, M.D.; Morris, M.J.; Crona, D.J.; George, D.J.; Dreicer, R.; Tse, K.; Petruck, J.; Webb, I.J.; Bander, N.H.; et al. Phase 1/2 multiple ascending dose trial of the prostate-specific membrane antigen-targeted antibody drug conjugate MLN2704 in metastatic castration-resistant prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2016, 34, 530.e15–530.e21. [Google Scholar] [CrossRef] [Green Version]
- Mita, M.M.; Ricart, A.D.; Mita, A.C.; Patnaik, A.; Sarantopoulos, J.; Sankhala, K.; Fram, R.J.; Qin, A.; Watermill, J.; Tolcher, A.W.; et al. A phase I study of a CanAg-targeted immunoconjugate, huC242-DM4, in patients with Can Ag-expressing solid tumors. J. Clin. Oncol. 2007, 25, 3062. [Google Scholar] [CrossRef]
- Borghaei, H.; O’Malley, D.M.; Seward, S.M.; Bauer, T.M.; Perez, R.P.; Oza, A.M.; Jeong, W.; Michenzie, M.F.; Kirby, M.W.; Chandorkar, G.; et al. Phase 1 study of IMGN853, a folate receptor alpha (FRα)-targeting antibody-drug conjugate (ADC) in patients (Pts) with epithelial ovarian cancer (EOC) and other FRA-positive solid tumors. J. Clin. Oncol. 2015, 33, 5558. [Google Scholar] [CrossRef]
- Younes, A.; Kim, S.; Romaguera, J.; Copeland, A.; Farial Sde, C.; Kwak, L.W.; Fayad, L.; Hagemeister, F.; Fanale, M.; Neelapu, S.; et al. Phase I multidose-escalation study of the anti-CD19 maytansinoid immunoconjugate SAR3419 administered by intravenous infusion every 3 weeks to patients with relapsed/refractory B-cell lymphoma. J. Clin. Oncol. 2012, 30, 2776–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, J.A.; Motzer, R.; Molina, A.M.; Choueiri, T.K.; Heath, E.I.; Kollmannsberger, C.K.; Redman, B.G.; Sangha, R.S.; Ernst, D.S.; Pili, R.; et al. Phase I studies of anti-ENPP3 antibody drug conjugates (ADCs) in advanced refractory renal cell carcinomas (RRCC). J. Clin. Oncol. 2015, 33, 2503. [Google Scholar] [CrossRef]
- Tannir, N.M.; Forero-Torres, A.; Ramchandren, R.; Pal, S.K.; Ansell, S.M.; Infante, J.R.; de Vos, S.; Hamlin, P.A.; Kim, S.K.; Whiting, N.C.; et al. Phase I dose-escalation study of SGN-75 in patients with CD70-positive relapsed/refractory non-Hodgkin lymphoma or metastatic renal cell carcinoma. Investig. New Drugs 2014, 32, 1246–1257. [Google Scholar] [CrossRef]
- Eaton, J.S.; Miller, P.E.; Mannis, M.J.; Murphy, C.J. Ocular Adverse Events Associated with Antibody–Drug Conjugates in Human Clinical Trials. J. Ocul. Pharmacol. 2015, 31, 589–604. [Google Scholar] [CrossRef]
- Hong, D.S.; Concin, N.; Vergote, I.; de Bono, J.S.; Slomovitz, B.M.; Drew, Y.; Arkenau, H.-T.; Machiels, J.-P.; Spicer, J.F.; Jones, R.; et al. Tisotumab Vedotin in Previously Treated Recurrent or Metastatic Cervical Cancer. Clin. Cancer Res. 2020, 26, 1220–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Goeij, B.E.; Lambert, J.M. New developments for antibody-drug conjugate-based therapeutic approaches. Curr. Opin. Immunol. 2016, 40, 14–23. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.T.; Chen, Y.; Marhoul, J.; Jacobson, F. Statistical Modeling of the Drug Load Distribution on Trastuzumab Emtansine (Kadcyla), a Lysine-Linked Antibody Drug Conjugate. Bioconjugate Chem. 2014, 25, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef]
- Matsuda, Y.; Mendelsohn, B.A. Recent Advances in Drug–Antibody Ratio Determination of Antibody–Drug Conjugates. Chem. Pharm. Bull. 2021, 69, 976–983. [Google Scholar] [CrossRef] [PubMed]
- Junutula, J.R.; Raab, H.; Clark, S.; Bhakta, S.; Leipold, D.D.; Weir, S.; Chen, Y.; Simpson, M.; Tsai, S.P.; Dennis, M.S.; et al. Site-specific conjugation of a cytotoxic drug to an antibody improves the therapeutic index. Nat. Biotechnol. 2008, 26, 925–932. [Google Scholar] [CrossRef]
- Herrera, A.F.; Patel, M.R.; Burke, J.M.; Advani, R.H.; Cheson, B.D.; Sharman, J.P.; Penuel, E.; Polson, A.G.; Leng, N.; Li, C.; et al. A Phase I Study of the Anti-CD79b THIOMABTM-Drug Conjugate DCDS0780A in Patients (pts) with Relapsed or Refractory B-Cell Non-Hodgkin’s Lymphoma (B-NHL). Blood 2017, 130, 4129. [Google Scholar] [CrossRef]
- Drake, P.M.; Albers, A.E.; Baker, J.; Banas, S.; Barfield, R.M.; Bhat, A.S.; de Hart, G.W.; Garofalo, A.W.; Holder, P.; Jones, L.C.; et al. Aldehyde Tag Coupled with HIPS Chemistry Enables the Production of ADCs Conjugated Site-Specifically to Different Antibody Regions with Distinct in Vivo Efficacy and PK Outcomes. Bioconjugate Chem. 2014, 25, 1331–1341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, P.; Bertozzi, C.R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015, 26, 176–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, S.J.; Bargh, J.D.; Dannheim, F.M.; Hanby, A.R.; Seki, H.; Counsell, A.J.; Ou, X.; Fowler, E.; Ashman, N.; Takada, Y.; et al. Site-selective modification strategies in antibody–drug conjugates. Chem. Soc. Rev. 2021, 50, 1305–1353. [Google Scholar] [CrossRef]
- Schumacher, D.; Hackenberger, C.P.R.; Leonhardt, H.; Helma, J. Current Status: Site-Specific Antibody Drug Conjugates. J. Clin. Immunol. 2016, 36, 100–107. [Google Scholar] [CrossRef] [Green Version]
- Behrens, C.R.; Liu, B. Methods for site-specific drug conjugation to antibodies. mAbs 2014, 6, 46–53. [Google Scholar] [CrossRef] [Green Version]
- Matsuda, Y.; Mendelsohn, B.A. An overview of process development for antibody-drug conjugates produced by chemical conjugation technology. Expert Opin. Biol. Ther. 2021, 21, 963–975. [Google Scholar] [CrossRef]
- Hussain, A.F.; Grimm, A.; Sheng, W.; Zhang, C.; Al-Rawe, M.; Bräutigam, K.; Abu Mraheil, M.; Zeppernick, F.; Meinhold-Heerlein, I. Toward Homogenous Antibody Drug Conjugates Using Enzyme-Based Conjugation Approaches. Pharmaceuticals 2021, 14, 343. [Google Scholar] [CrossRef]
- Burke, P.J.; Hamilton, J.Z.; Jeffrey, S.C.; Hunter, J.H.; Doronina, S.O.; Okeley, N.M.; Miyamoto, J.B.; Anderson, M.E.; Stone, I.J.; Ulrich, M.L.; et al. Optimization of a PEGylated Glucuronide-Monomethylauristatin E Linker for Antibody–Drug Conjugates. Mol. Cancer Ther. 2017, 16, 116–123. [Google Scholar] [CrossRef] [Green Version]
- Simmons, J.K.; Burke, P.J.; Cochran, J.H.; Pittman, P.G.; Lyon, R.P. Reducing the antigen-independent toxicity of antibody-drug conjugates by minimizing their non-specific clearance through PEGylation. Toxicol. Appl. Pharmacol. 2020, 392, 114932. [Google Scholar] [CrossRef]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C.; et al. Synthesis and Evaluation of Hydrophilic Linkers for Antibody–Maytansinoid Conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef]
- Hartimath, S.V.; Alizadeh, E.; Solomon, V.R.; Chekol, R.; Bernhard, W.; Hill, W.; Parada, A.C.; Barreto, K.; Geyer, C.R.; Fonge, H.; et al. Preclinical Evaluation of111In-Labeled PEGylated Maytansine Nimotuzumab Drug Conjugates in EGFR-Positive Cancer Models. J. Nucl. Med. 2019, 60, 1103–1110. [Google Scholar] [CrossRef] [Green Version]
- Shao, S.; Tsai, M.-H.; Lu, J.; Yu, T.; Jin, J.; Xiao, D.; Jiang, H.; Han, M.; Wang, M.; Wang, J. Site-specific and hydrophilic ADCs through disulfide-bridged linker and branched PEG. Bioorganic Med. Chem. Lett. 2018, 28, 1363–1370. [Google Scholar] [CrossRef]
- Cheng, X.; Li, J.; Tanaka, K.; Majumder, U.; Milinichik, A.Z.; Verdi, A.C.; Maddage, C.J.; Rybinski, K.A.; Fernando, S.; Fernando, D.; et al. MORAb-202, an Antibody–Drug Conjugate Utilizing Humanized Anti-human FRα Farletuzumab and the Microtubule-targeting Agent Eribulin, has Potent Antitumor Activity. Mol. Cancer Ther. 2018, 17, 2665–2675. [Google Scholar] [CrossRef] [Green Version]
- Kovtun, Y.; Noordhuis, P.; Whiteman, K.R.; Watkins, K.; Jones, G.E.; Harvey, L.; Lai, K.C.; Portwood, S.; Adams, S.; Sloss, C.M.; et al. IMGN779, a Novel CD33-Targeting Antibody–Drug Conjugate with DNA-Alkylating Activity, Exhibits Potent Antitumor Activity in Models of AML. Mol. Cancer Ther. 2018, 17, 1271–1279. [Google Scholar] [CrossRef] [Green Version]
- Tiberghien, A.C.; Levy, J.-N.; Masterson, L.A.; Patel, N.V.; Adams, L.R.; Corbett, S.; Williams, D.G.; Hartley, J.A.; Howard, P.W. Design and Synthesis of Tesirine, a Clinical Antibody–Drug Conjugate Pyrrolobenzodiazepine Dimer Payload. ACS Med. Chem. Lett. 2016, 7, 983–987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flynn, M.J.; Zammarchi, F.; Tyrer, P.C.; Akarca, A.U.; Janghra, N.; Britten, C.E.; Havenith, C.E.G.; Levy, J.-N.; Tiberghien, A.; Masterson, L.A.; et al. ADCT-301, a Pyrrolobenzodiazepine (PBD) Dimer–Containing Antibody–Drug Conjugate (ADC) Targeting CD25-Expressing Hematological Malignancies. Mol. Cancer Ther. 2016, 15, 2709–2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandall, S.L.; Mason, M.; Olson, D.; Mazahreh, R.; Pires, T.; Sahetya, D.; Westendorf, L.; Leiske, C.; Schimpf, B.; Nguyen, L.; et al. SGN-CD228A: A novel humanized anti-CD228 antibody-drug conjugate for the treatment of solid tumors. Cancer Res. 2019, 79, 2688. [Google Scholar] [CrossRef]
- Syed, Y.Y. Sacituzumab Govitecan: First Approval. Drugs 2020, 80, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Carl, P.L.; Chakravarty, P.K.; Katzenellenbogen, J.A. A novel connector linkage applicable in prodrug design. J. Med. Chem. 1981, 24, 479–480. [Google Scholar] [CrossRef]
- Edupuganti, V.V.S.R.; Tyndall, J.D.A.; Gamble, A.B. Self-immolative Linkers in Prodrugs and Antibody Drug Conjugates in Cancer Treatment. Recent Pat. Anti-Cancer Drug Discov. 2021, 16, 479–497. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody-Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef]
- Pillow, T.H.; Schutten, M.; Yu, S.-F.; Ohri, R.; Sadowsky, J.; Poon, K.A.; Solis, W.; Zhong, F.; del Rosario, G.; Go, M.A.T.; et al. Modulating Therapeutic Activity and Toxicity of Pyrrolobenzodiazepine Antibody–Drug Conjugates with Self-Immolative Disulfide Linkers. Mol. Cancer Ther. 2017, 16, 871–878. [Google Scholar] [CrossRef] [Green Version]
- Chuprakov, S.; Ogunkoya, A.O.; Barfield, R.M.; Bauzon, M.; Hickle, C.; Kim, Y.C.; Yeo, D.; Zhang, F.; Rabuka, D.; Drake, P.M. Tandem-Cleavage Linkers Improve the In Vivo Stability and Tolerability of Antibody–Drug Conjugates. Bioconjugate Chem. 2021, 32, 746–754. [Google Scholar] [CrossRef]
- Anami, Y.; Yamazaki, C.M.; Xiong, W.; Gui, X.; Zhang, N.; An, Z.; Tsuchikama, K. Glutamic acid–valine–citrulline linkers ensure stability and efficacy of antibody–drug conjugates in mice. Nat. Commun. 2018, 9, 2512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, S.Y.Y.; Anami, Y.; Yamazaki, C.M.; Xiong, W.; Haase, C.M.; Olson, S.D.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; et al. An Enzymatically Cleavable Tripeptide Linker for Maximizing the Therapeutic Index of Antibody–Drug Conjugates. Mol. Cancer Ther. 2022, 21, 1449–1461. [Google Scholar] [CrossRef]
- Miller, M.L.; Fishkin, N.E.; Li, W.; Whiteman, K.R.; Kovtun, Y.; Reid, E.E.; Archer, K.E.; Maloney, E.K.; Audette, C.A.; Mayo, M.F.; et al. A New Class of Antibody-Drug Conjugates with Potent DNA Alkylating Activity. Mol. Cancer Ther. 2016, 15, 1870–1878. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Reid, E.E.; Harris, L.; Salomon, P.L.; Miller, M.L.; Chari, R.V.J.; Keating, T.A. Antibody–Drug Conjugates with Indolinobenzodiazepine Dimer Payloads: DNA-Binding Mechanism of Indolinobenzodiazepine Dimer Catabolites in Target Cancer Cells. Mol. Pharmaceutics 2020, 17, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.L.; Shizuka, M.; Wilhelm, A.; Salomon, P.; Reid, E.E.; Lanieri, L.; Sikka, S.; Maloney, E.K.; Harvey, L.; Qiu, Q.; et al. A DNA-Interacting Payload Designed to Eliminate Cross-Linking Improves the Therapeutic Index of Antibody–Drug Conjugates (ADCs). Mol. Cancer Ther. 2018, 17, 650–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damelin, M.; Zhong, W.; Myers, J.; Sapra, P. Evolving Strategies for Target Selection for Antibody-Drug Conjugates. Pharm. Res. 2015, 32, 3494–3507. [Google Scholar] [CrossRef]
- Lin, J.; Sagert, J. Targeting Drug Conjugates to the Tumor Microenvironment: Probody Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin, M., Ed.; Springer International Publishing: Cham, Germany, 2018; pp. 281–298. [Google Scholar]
- Kessenbrock, K.; Plaks, V.; Werb, Z. Matrix Metalloproteinases: Regulators of the Tumor Microenvironment. Cell 2010, 141, 52–67. [Google Scholar] [CrossRef] [Green Version]
- Sevenich, L.; Joyce, J.A. Pericellular proteolysis in cancer. Genes Dev. 2014, 28, 2331–2347. [Google Scholar] [CrossRef] [Green Version]
- Fujimori, K.; Covell, D.G.; Fletcher, J.E.; Weinstein, J.N. A modeling analysis of monoclonal antibody percolation through tumors: A binding-site barrier. J. Nucl. Med. 1990, 31, 1191–1198. [Google Scholar]
- Bordeau, B.M.; Yang, Y.; Balthasar, J.P. Transient Competitive Inhibition Bypasses the Binding Site Barrier to Improve Tumor Penetration of Trastuzumab and Enhance T-DM1 Efficacy. Cancer Res. 2021, 81, 4145–4154. [Google Scholar] [CrossRef]
- Bordeau, B.M.; Abuqayyas, L.; Nguyen, T.D.; Chen, P.; Balthasar, J.P. Development and Evaluation of Competitive Inhibitors of Trastuzumab-HER2 Binding to Bypass the Binding-Site Barrier. Front. Pharm. 2022, 13, 837744. [Google Scholar] [CrossRef]
- Chen, P.; Bordeau, B.M.; Zhang, Y.; Balthasar, J.P. Transient Inhibition of Trastuzumab-Tumor Binding to Overcome the “Binding-Site Barrier” and Improve the Efficacy of a Trastuzumab-Gelonin Immunotoxin. Mol. Cancer 2022, 21, 1573–1582. [Google Scholar] [CrossRef] [PubMed]
- Sagert, J.G.; West, J.W.; Wong, C.; Desnoyers, L.R.; Vasiljeva, O.; Richardson, J.H.; Polu, K.R.; Lowman, H.B. Abstract 2665: Transforming Notch ligands into tumor-antigen targets: A Probody-Drug Conjugate (PDC) targeting Jagged 1 and Jagged 2. Cancer Res. 2014, 74, 2665. [Google Scholar] [CrossRef]
- Autio, K.A.; Boni, V.; Humphrey, R.W.; Naing, A. Probody Therapeutics: An Emerging Class of Therapies Designed to Enhance On-Target Effects with Reduced Off-Tumor Toxicity for Use in Immuno-Oncology. Clin. Cancer Res. 2020, 26, 984–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Corbacho, J.; Spira, A.; Boni, V.; Feliu, J.; Middleton, M.; Burris, H.; Weaver, A.; Will, M.; Harding, J.; Meric-Bernstam, F.; et al. 422TiPPROCLAIM-CX-2009: A first-in-human trial to evaluate CX-2009 in adults with metastatic or locally advanced unresectable solid tumors. Ann. Oncol. 2017, 28, V140. [Google Scholar] [CrossRef] [Green Version]
- Johnson, M.; El-Khoueiry, A.; Hafez, N.; Lakhani, N.; Mamdani, H.; Rodon, J.; Sanborn, R.E.; Garcia-Corbacho, J.; Boni, V.; Stroh, M.; et al. Phase I, First-in-Human Study of the Probody Therapeutic CX-2029 in Adults with Advanced Solid Tumor Malignancies. Clin. Cancer Res. 2021, 27, 4521–4530. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Shen, J.; Yu, W.; Jing, C.; Saha, S.; Rangan, V.; Richardson, J.; Liu, Y.; Boule, S.; Lucas, J.; et al. CX-2043, an EpCAM-targeting probody drug conjugate, demonstrates anti-tumor activity with a favorable safety profile in preclinical models. Eur. J. Cancer 2020, 138, S15. [Google Scholar] [CrossRef]
- Comer, F.; Gao, C.; Coats, S. Bispecific and Biparatopic Antibody Drug Conjugates. In Innovations for Next-Generation Antibody-Drug Conjugates; Damelin, M., Ed.; Springer International Publishing: Cham, Germany, 2018; pp. 267–280. [Google Scholar]
- May, C.; Sapra, P.; Gerber, H.-P. Advances in bispecific biotherapeutics for the treatment of cancer. Biochem. Pharmacol. 2012, 84, 1105–1112. [Google Scholar] [CrossRef]
- Mazor, Y.; Oganesyan, V.; Yang, C.; Hansen, A.; Wang, J.; Liu, H.; Sachsenmeier, K.; Carlson, M.; Gadre, D.V.; Borrok, M.J.; et al. Improving target cell specificity using a novel monovalent bispecific IgG design. mAbs 2015, 7, 377–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, Y.; Hansen, A.; Yang, C.; Chowdhury, P.S.; Wang, J.; Stephens, G.; Wu, H.; Dall’Acqua, W.F. Insights into the molecular basis of a bispecific antibody’s target selectivity. mAbs 2015, 7, 461–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazor, Y.; Sachsenmeier, K.F.; Yang, C.; Hansen, A.; Filderman, J.; Mulgrew, K.; Wu, H.; Dall’Acqua, W.F. Enhanced tumor-targeting selectivity by modulating bispecific antibody binding affinity and format valence. Sci. Rep. 2017, 7, 40098. [Google Scholar] [CrossRef] [PubMed]
- Sellmann, C.; Doerner, A.; Knuehl, C.; Rasche, N.; Sood, V.; Krah, S.; Rhiel, L.; Messemer, A.; Wesolowski, J.; Schuette, M.; et al. Balancing Selectivity and Efficacy of Bispecific Epidermal Growth Factor Receptor (EGFR) × c-MET Antibodies and Antibody-Drug Conjugates. J. Biol. Chem. 2016, 291, 25106–25119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giovannini, M.; Gregorc, V.; Belli, C.; Roca, E.; Lazzari, C.; Viganò, M.G.; Serafico, A.; Villa, E. Clinical Significance of Skin Toxicity due to EGFR-Targeted Therapies. J. Oncol. 2009, 2009, 849051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holcmann, M.; Sibilia, M. Mechanisms underlying skin disorders induced by EGFR inhibitors. Mol. Cell. Oncol. 2015, 2, e1004969. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, A.; Anami, Y.; Ha, S.Y.Y.; Roeder, T.J.; Xiong, W.; Lee, J.; Ueno, N.T.; Zhang, N.; An, Z.; Tsuchikama, K. Chemical generation of small molecule-based bispecific antibody-drug conjugates for broadening the target scope. Bioorganic Med. Chem. 2021, 32, 116013. [Google Scholar] [CrossRef] [PubMed]
- De Goeij, B.E.C.G.; Vink, T.; Ten Napel, H.; Breij, E.C.W.; Satijn, D.; Wubbolts, R.; Miao, D.; Parren, P.W.H.I. Efficient Payload Delivery by a Bispecific Antibody–Drug Conjugate Targeting HER2 and CD63. Mol. Cancer Ther. 2016, 15, 2688–2697. [Google Scholar] [CrossRef] [Green Version]
- Andreev, J.; Thambi, N.; Perez Bay, A.E.; Delfino, F.; Martin, J.; Kelly, M.P.; Kirshner, J.R.; Rafique, A.; Kunz, A.; Nittoli, T.; et al. Bispecific Antibodies and Antibody–Drug Conjugates (ADCs) Bridging HER2 and Prolactin Receptor Improve Efficacy of HER2 ADCs. Mol. Cancer Ther. 2017, 16, 681–693. [Google Scholar] [CrossRef] [Green Version]
- Sapra, P.; Betts, A.; Boni, J. Preclinical and clinical pharmacokinetic/pharmacodynamic considerations for antibody-drug conjugates. Expert Rev. Clin. Pharm. 2013, 6, 541–555. [Google Scholar] [CrossRef]
- Smith, T.W.; Haber, E.; Yeatman, L.; Butler, V.P., Jr. Reversal of advanced digoxin intoxication with Fab fragments of digoxin-specific antibodies. N. Engl. J. Med. 1976, 294, 797–800. [Google Scholar] [CrossRef] [PubMed]
- Pollack, C.V.; Reilly, P.A.; Eikelboom, J.; Glund, S.; Verhamme, P.; Bernstein, R.A.; Dubiel, R.; Huisman, M.V.; Hylek, E.M.; Kamphuisen, P.W.; et al. Idarucizumab for Dabigatran Reversal. N. Engl. J. Med. 2015, 373, 511–520. [Google Scholar] [CrossRef] [Green Version]
- Balthasar, J.; Fung, H.L. Utilization of antidrug antibody fragments for the optimization of intraperitoneal drug therapy: Studies using digoxin as a model drug. J. Pharmacol. Exp. Ther. 1994, 268, 734–739. [Google Scholar]
- Balthasar, J.P.; Fung, H.L. Inverse targeting of peritoneal tumors: Selective alteration of the disposition of methotrexate through the use of anti-methotrexate antibodies and antibody fragments. J. Pharm. Sci. 1996, 85, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Soda, D.M.; Balthasar, J.P. Application of pharmacokinetic-pharmacodynamic modeling to predict the kinetic and dynamic effects of anti-methotrexate antibodies in mice. J. Pharm. Sci. 2003, 92, 1665–1676. [Google Scholar] [CrossRef] [PubMed]
- Lobo, E.D.; Balthasar, J.P. Application of anti-methotrexate Fab fragments for the optimization of intraperitoneal methotrexate therapy in a murine model of peritoneal cancer. J. Pharm. Sci. 2005, 94, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.K.; Balthasar, J.P. PK/TD modeling for prediction of the effects of 8C2, an anti-topotecan mAb, on topotecan-induced toxicity in mice. Int. J. Pharm. 2014, 465, 228–238. [Google Scholar] [CrossRef] [Green Version]
- Bordeau, B.M.; Nguyen, T.D.; Polli, R.; Chen, P.; Balthasar, J.P. Payload-binding fab fragments increase the therapeutic index of MMAE antibody-drug conjugates. Mol. Cancer Ther. 2023, accepted. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Use of Payload Binding Selectivity Enhancers to Improve Therapeutic Index of Maytansinoid-Antibody-Drug Conjugates. Mol. Cancer Ther. 2023, submitted.
Figure 1.
Mechanisms of ADC toxicity. Uptake of intact ADCs into normal cells may occur through non-specific endocytosis, or through internalization upon binding to the target antigen or to Fc/C-type lectin receptors. Payloads released from ADC deconjugation or other targeted/non-targeted apoptotic cells in the extracellular fluid may also enter normal cells via passive diffusion for membrane-permeable payloads or via non-specific endocytosis for membrane-impermeable linker-payload adducts. Created with BioRender.com.

Table 2.
Frequently reported dose-limiting toxicities associated with payload classes.
| Approved/Late-Stage ADCs | Linker Type | Key Toxicities | |
|---|---|---|---|
| Tubulin inhibitors | |||
| MMAE | Brentuximab Vedotin, Polatuzumab Vedotin, Enfortumab Vedotin, Tisotumab Vedotin, Disitamab Vedotin | Cleavable | Neutropenia, peripheral neuropathy, anemia, skin toxicity |
| MMAF | Belantamab Mafodotin | Non-cleavable | Thrombocytopenia, ocular toxicity, hepatic toxicity |
| DM1 | Trastuzumab Emtansine | Non-cleavable | Thrombocytopenia, hepatic toxicity |
| DM4 | Mirvetuximab Soravtansine | Cleavable | Neutropenia, anemia, peripheral neuropathy, ocular toxicity |
| DNA-crosslinkers/ DNA-alkylators | |||
| Calicheamicin | Gemtuzumab Ozogamicin, Inotuzumab Ozogamicin | Cleavable | Neutropenia, thrombocytopenia, hepatic toxicity |
| PBD | Loncastuximab Tesirine | Cleavable | Neutropenia, thrombocytopenia, anemia, serosal effusion, nephron toxicity, skin toxicity |
| Duocarmycin | Trastuzumab Duocarmazine | Cleavable | Neutropenia, thrombocytopenia, serosal effusion |
| Topoisomerase inhibitors | |||
| SN-38 | Sacituzumab Govitecan | Cleavable | Neutropenia, gastrointestinal toxicity |
| Deruxtecan | Trastuzumab Deruxtecan | Cleavable | Neutropenia, gastrointestinal toxicity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Nguyen, T.D.; Bordeau, B.M.; Balthasar, J.P. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers 2023, 15, 713. https://doi.org/10.3390/cancers15030713
AMA Style
Nguyen TD, Bordeau BM, Balthasar JP. Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers. 2023; 15(3):713. https://doi.org/10.3390/cancers15030713
Chicago/Turabian StyleNguyen, Toan D., Brandon M. Bordeau, and Joseph P. Balthasar. 2023. "Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability" Cancers 15, no. 3: 713. https://doi.org/10.3390/cancers15030713
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.

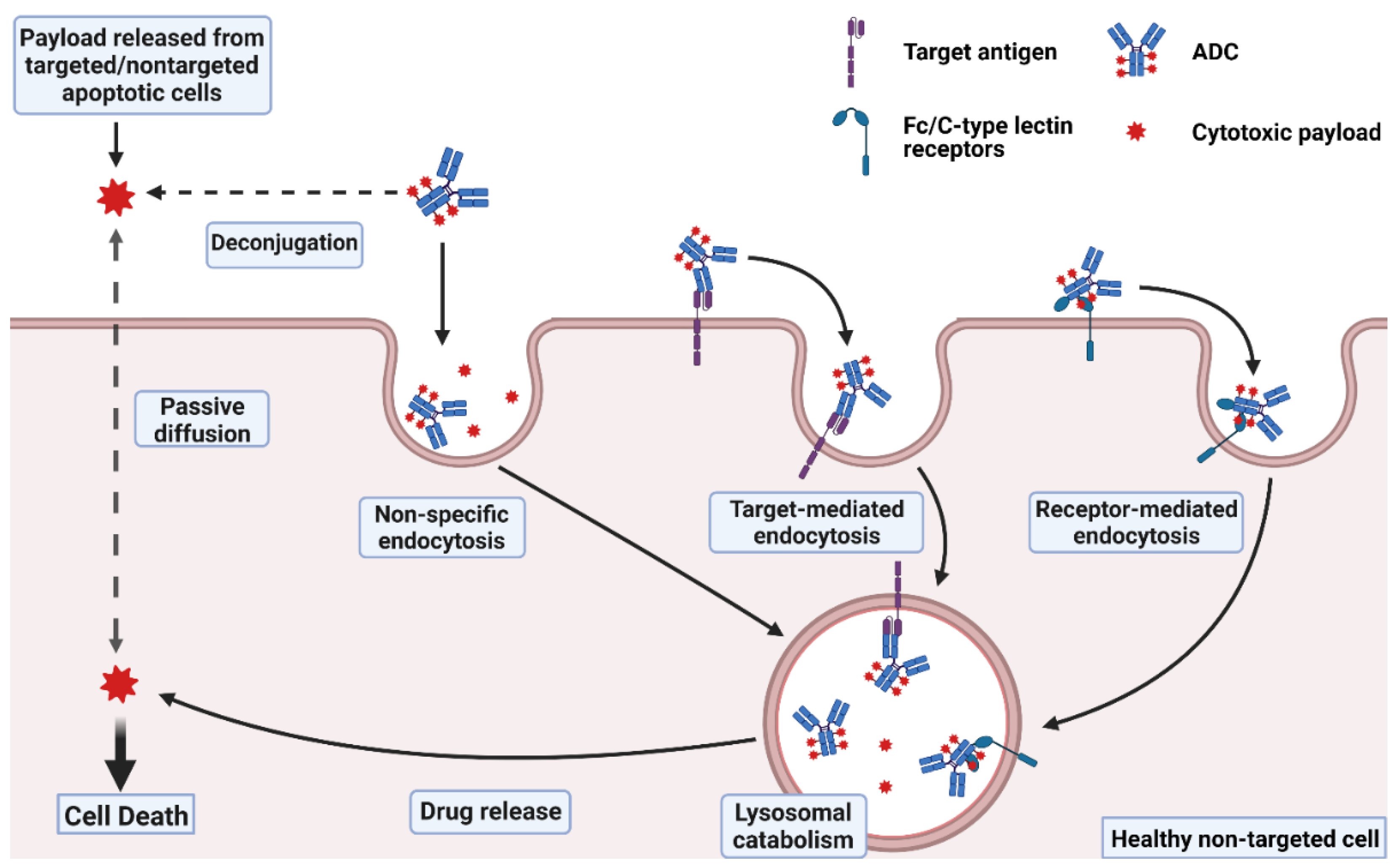{kind=link}