

摘要
针对细胞靶点筛选具有潜在生物活性的新化合物对药物发现和化学安全至关重要。转录组学提供了一种评估全局基因表达变化的有效方法,但从这些数据中解读化学机制往往具有挑战性。根据许多生物过程都与独特的基因表达特征(基因特征)相关联这一观察结果,连接性图谱是将化学品与机理联系起来的潜在数据驱动途径。然而,由于转录组数据包含数以千计的嘈杂基因,因此挖掘化学物质对基因特征对生物机制的影响具有挑战性。由于有望从不断增长的转录组数据中发现化学机制、新药和疾病靶点,人们不断开发新的连通性图谱方法,以区分信号和噪音。在此,我们将从不同的转录组技术、公共数据库、基因特征、模式匹配算法和统计评估标准等方面对这些方法进行分析。为了应对连通性图谱的复杂性,我们提出了一个统一的方案,以连贯地组织和比较已发表的工作流程。首先,我们基于微阵列、RNA-Seq 和 L1000 等各种转录组技术,对转录组图谱和基因特征的基本概念进行了标准化,并讨论了基因表达总库(GEO)、ArrayExpress 和 MSigDB 等广泛使用的数据源。接下来,我们将连接性映射概括为一种模式匹配任务,用于发现查询(如新化学物质的转录组概况)与参考(如已知目标的基因特征)之间的相似性。 已发表的模式匹配方法分为两大类:基于向量的使用相关性、雅卡指数等指标,以及基于聚合的使用参数和非参数统计(如基因组富集分析)。我们介绍了评估不同方法性能的统计方法,以及文献中报告的基准转录组数据集的比较。最后,我们回顾了连通性图谱在毒理学中的应用,并为利用浓度反应转录组数据评估化学物质诱导的毒性提供了指导。除了作为理解和实施连通性图谱工作流程的高级指南和教程外,我们还希望这篇综述能激发使用转录组数据评估化学安全性和药物发现的新算法。
关键词:化学筛选、基因表达、生物活性指纹、转录组剖析、基因特征、相似性算法、基因组富集、转录组浓度-反应、新方法方法
图表摘要
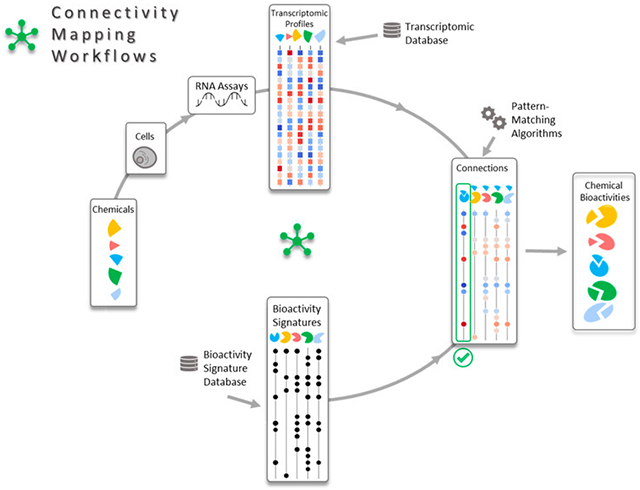
导言
药物发现和化学品安全需要有效的工具来筛选新化合物,以确定其针对细胞靶点的潜在生物活性。转录组学是通过影响全局基因表达来评估化学物质生物效应的广泛应用技术之一。1由于化学物质通过直接与受体结合2或间接破坏细胞稳态3来诱导基因表达变化,因此从转录组数据中推断其靶标具有挑战性。连接性图谱利用基因的 "通用语言 "4,5测量转录组图谱与细胞靶标相关基因特征之间的相似性,从而解决了这一问题。转录组学在人类基因组测序后的二十年间发展迅速。6从 cDNA 点阵7开始,到高密度寡核苷酸阵8,再到最近的 RNA 测序技术9 ,转录组学的可重复性、可靠性和成本效益不断提高。10因此,公共领域的资料库11、12现已提供了数以百万计的转录组图谱,涉及数千种疾病。4、13需要创新工具来利用这些丰富的转录组数据揭示化学品、途径和疾病之间的新关系。连接性图谱4就是这样一种工具,它可以促进药物发现14,帮助现有药物的再利用15,并生产出更安全的化学品。16
Connectivity mapping with transcriptomic data is one of many techniques in a rich landscape of computational methods for inferring the putative interactions between chemicals and biological targets or pathways. This landscape can be broadly divided into approaches based on binding, similarity, and machine learning (ML). Binding-based methods attempt to model physico-chemical interactions between a chemical and a protein target with three-dimensional structure data using molecular dynamics17,18 or, more recently, using ML.19 There have been impressive advances predicting new ligands for specific protein targets,20 and with predicted three-dimensional structures for all known proteins,21 virtually screening all chemicals against thousands of protein targets could be within reach.22
Connectivity mapping is conceptually related to other similarity-based approaches, which attempt to infer the properties of a new chemical using pair-wise similarity with chemicals of known properties, including physico-chemical properties or biological activities. If two chemicals have significant structural similarities, then similarity-based approaches assume they also have similar properties. Similarity-based approaches have two essential ingredients: a vector of attributes and a measure of similarity based on the attributes. Similarity-based pattern-matching techniques are also considered instance-based learning methods23 in ML, which includes approaches like k-nearest neighbor (KNN) classification. Chemical similarity-based approaches use molecular structure descriptors (such as extended connectivity fingerprints24) to represent chemicals and measure similarity using set operations (for a review of similarity measures, see Bero et al.25). For example, a query chemical can be searched against a database to find other structurally similar chemicals from which the unknown biological role can be inferred. Chemical structure-based similarity is widely used to infer molecular targets.26 One of the problems with using chemical similarity-based techniques is that minor alterations in structure can lead to drastic changes in their affinity for the same target, which are known as “activity cliffs” in structure-activity relationship (SAR) research.27 Another issue is that new structural categories of chemicals can be discovered or synthesized that have no existing analogues. If they bear insufficient resemblance to known chemicals, it is not possible to infer their properties based on structural similarity alone. Despite these limitations, structure-based automated prediction approaches28 are routinely used to fill data gaps for untested chemicals based on the known properties of analogues in the same local domains. More recently, structural and bioactivity similarity between chemicals has been used to infer the toxicity of untested chemicals.29–32
Finding pair-wise similarities using biological and chemical descriptors is a practical strategy for inferring the properties of untested chemicals; however, if hundreds of chemicals are associated with different classes of biological activities (e.g., protein target, pathway activation, toxicity, etc.), then ML can be more effective. ML algorithms systematically mine patterns in data (i.e., vector representations of data derived from biological and chemical descriptors) to build accurate predictive models of various biological activities.33 For example, ML algorithms mine chemical structure representations to build models, referred to as quantitative structure-activity relations (QSARs).34 QSAR models have been used to classify potential nuclear receptor activators,35,36 cellular stress responses,37,38 and toxicities.39,40 Similarly, ML algorithms mine transcriptomic data on chemicals (derived from different cellular contexts) to build models of biological mechanisms,41–44 and toxicities.45,46 Models derived by ML can predict the bioactivity or toxicity of new or untested chemicals using vector representations of data (i.e. attribute-value vectors that are used to train the model). Different ML methods have varying requirements for training data to produce reliable predictive models. Whereas similarity-based approaches such as KNN may only require a few examples because of their simplicity, more complex ML algorithms need varying amounts of training data to tune model parameters reliably. Furthermore, for in vivo toxicity prediction, it is also essential to consider the chemical dose, duration, and route of exposure. A systematic comparison of similarity-based and other ML algorithms is beyond the scope of this review.
Connectivity mapping may be considered an automated biological read-across47,48 technique to infer properties of untested substances using transcriptomic profiles in place of chemical structure representations. Gene-based descriptors in transcriptomic profiles measure the expression of specific genes in the genome, just like structure descriptors capture the presence of substructural moieties in chemicals. Transcriptomic profiles, however, can capture the biological response to chemical treatments, genetic perturbations, or pathological conditions using continuous expression levels of genes in ways that chemical structure descriptors cannot. The ability of transcriptomics to capture a diverse array of physiological states also makes it a powerful tool for finding similarity-based connections. This review is a guide for navigating connectivity mapping in terms of the diverse array of technologies to generate transcriptomic profiles, define biological states using gene-based descriptors, and organize the plethora of algorithms to measure transcriptomic similarity.
Historical Background
Connectivity mapping originates from functional discovery studies,49 which aimed to interpret the molecular phenotypes of biological samples using transcriptomics.50,51 A pivotal study by Hughes et al. produced one of the earliest and largest compendia of transcriptomic profiles for 300 genetic and chemical perturbations in yeast.52 The authors used similarity between transcriptomic profiles to cluster known mutants, uncharacterized mutants, and pharmacologic agents. For example, deleting YER044c, an uncharacterized yeast open reading frame (ORF), produced transcriptional profiles similar to the sterol isomerase (ERG2) deletion mutant. Further experiments determined that the YER044c ORF encoded the endoplasmic reticulum protein (ERG28). Because ERG2 and ERG28 are both involved in ergosterol biosynthesis, their deletion mutants produced similar transcriptomic profiles. Hughes et al. also showed transcriptomic responses to the drug fenpropimorph were similar to the responses due to ERG2 deletion mutants. This is not surprising as fenpropimorph is a fungicide that disrupts eukaryotic sterol biosynthesis pathways. Surprisingly, fenpropimorph was also a potent mammalian antagonist of sigma-1 receptor (SIGMAR1), which is involved in neuromodulatory pathways involved in pain. SIGMAR1 antagonists are being explored as a novel class of analgesic agents for treating pain.53 There is growing evidence that ERG2 disruptors in yeast are SIGMAR1 antagonists,54 and such pharmacological agents can be identified by connectivity mapping. The ability to link chemicals to mechanisms within and across species showed the value of transcriptomics as a “universal phenotype” for fingerprinting global biological states and of transcriptomic similarity to uncover novel relationships between chemicals and their targets.
Before connectivity mapping approaches, transcriptomics mainly identified differentially expressed genes between cases and controls using p-value and fold-change thresholds. Lists of differentially expressed genes helped identify statistically over-represented pathways (e.g., using Fisher’s Exact Test55) and provided insight into putative biological mechanisms (see Khatri and Draghici56, and Rivals et al.57)). However, because gene lists are sensitive to the choice of differential expression thresholds, using varying statistical cut-offs can produce inconsistent biological interpretations. Mootha et al. showed over-representation analysis of gene lists ignored the subtle yet coordinated regulation of gene sets relevant to a pathway. They found a gene set for the oxidative phosphorylation pathway “enriched” in diabetic versus healthy muscle tissues even though individual genes in the set were not significantly differentially expressed.58,59 Mootha et al.58 and Subramanian et al.59 called this approach gene set enrichment analysis (GSEA). Other gene set analysis (GSA) approaches subsequently used for pathway, and function enrichment60–62 have been reviewed extensively elsewhere.56,57
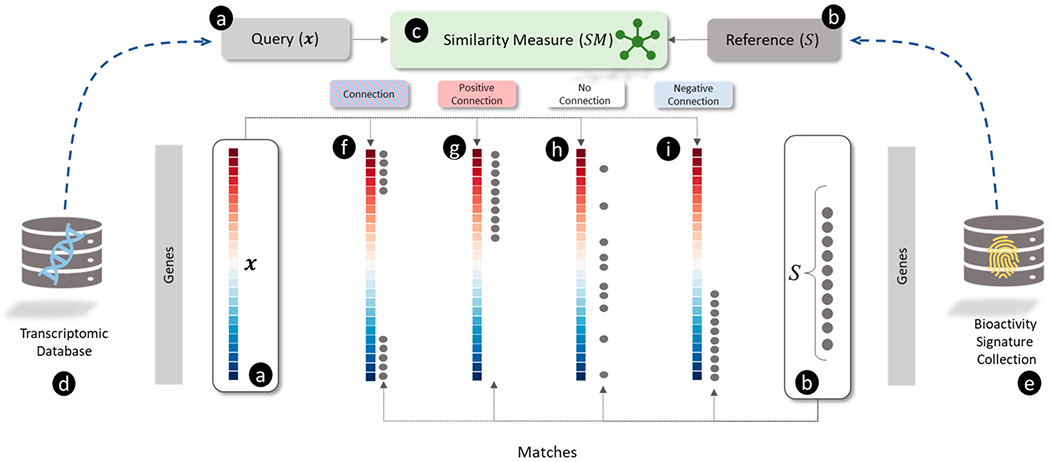
The connectivity map (CMap) project, which gave rise to the eponymous “connectivity mapping” approach, was the first publicly available large-scale compendium of transcriptomic profiles generated by treating human cells with a library of small molecules.5 Connectivity mapping used this compendium of 564 unique transcriptomic profiles for 164 chemicals (Build 01 of the CMap database, which we refer to as CMap v1). Connectivity, or similarity, was measured using a modified version of GSEA for analyzing “gene signatures” derived from highly up- and down-regulated genes in the transcriptomic profiles (see Figure 1). For example, Lamb et al.5 searched a signature of histone deacetylase (HDAC) inhibitors against the CMap v1 reference database. The HDAC inhibitor signature was derived from an independent study of HDAC inhibitors in bladder and breast cancer cells,63 which comprised eight up-regulated and five down-regulated genes (illustrated in Figure 1(a)). Searching the entire CMap v1 database (illustrated in Figure 1(e)) with this HDAC signature using GSEA (illustrated in Figure 1(c)), identified the most robust connections with vorinostat and trichostatin A (an example of such a match is shown in Figure (1(f)), both HDAC inhibitors. The ability of GSEA to link signatures of HDAC inhibitors from disparate experiments provided compelling evidence for the utility of connectivity mapping approaches.
Figure 1.
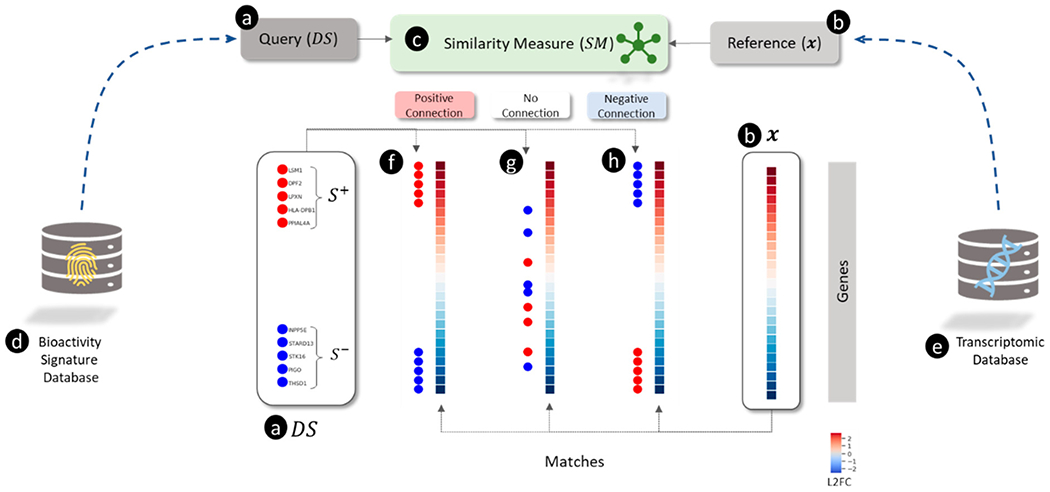
Overview of connectivity mapping as a pattern matching between (a) query and (b) reference using a (c) similarity measure illustrating different types of matches (f, g, and h). (a) The query is a directional gene signature (
In the second example, López et al. searched a gene signature of diet-induced obesity in rats.64 Using GSEA, they found a strong match between this signature and transcriptomic profiles for troglitazone, rosiglitazone, and indomethacin, all peroxisome proliferator-activated receptor gamma (PPARG) agonists. However, the directions of gene expression in the diet-induced obesity signature (i.e., up- and down-regulated genes) were found to be opposite to the directions of the genes in the profile for PPARG agonists. Such matches are referred to as “negative connections” as they have negative GSEA scores (see Figure 1(i) for a visual example of a negative connection). Interestingly, PPARG agonists are prescribed as hypolipidemic agents for the treatment of diabetes but can produce weight gain and liver injury as unwanted side effects. Thus, connectivity mapping revealed that the biological state of diet-induced obesity is “negatively connected” with PPARG-mediated hypolipidemic activity, notwithstanding differences in cells, treatment conditions, and gene expression assaying technologies. Finding negative connections between disease gene signatures and transcriptomic profiles of approved drugs forms the basis of some drug-repurposing approaches.15 These findings further demonstrated the utility of transcriptomic connectivity mapping for linking disease phenotypes with putative chemical treatments based on gene signatures. The initial success of connectivity mapping led to an expansion of the CMap (Build 02 of the CMap database, which we refer to as v2) to cover 1,309 chemicals and 6,100 transcriptomic profiles.65
Connectivity mapping and toxicology
A key challenge in toxicology is evaluating the safety of chemicals by determining their potency and potential for activating molecular targets that can lead to adverse health outcomes.66 In computational toxicology, transcriptomic profiling is used to rapidly screen thousands of untested chemicals to identify their putative targets, mechanism of action, or other effects.10,67–69 This is because high-throughput transcriptomic profiling using mRNA sequencing (RNA-Seq),9 and more recently targeted RNA-Seq,70 are extremely promising and cost-effective approaches for generating transcriptomic profiles for tens of thousands of chemical treatments. Whether evaluating new chemical entities for drug discovery or untested environmental chemicals for public health protection, transcriptomic connectivity mapping is a robust and high-throughput alternative to the existing techniques.16 Therefore, it is essential to examine the landscape of connectivity mapping approaches, understand their operation transparently, and assess their utility for specific toxicology applications.
Harmonizing connectivity mapping approaches
Dozens of refinements or alternatives to connectivity mapping have been proposed and are reviewed elsewhere.71,72 In this review, we develop a coherent view of various connectivity mapping approaches with an emphasis on three main ingredients: a transcriptomic profile produced by a perturbagen, a gene signature associated with a biological state, and an approach for matching the profile with the signature. The connectivity mapping workflow can be generalized as a database search and retrieval operation (see Figure 1) in which a “query” object (Figure (1a)) is compared with an extensive collection of “reference” (Figure 1(b)) objects (from a reference database (Figure 1(e))) using a pattern matching algorithm (Figure 1(c)) to find the most similar “hits” (Figure 1(f) and (h))). We employ the generic term “object” to cover several kinds of gene set-based inputs (summarized visually in Figure 2) for pattern matching. The variation between the connectivity mapping approaches is explained by the differences in the choice of the query, the reference database, and the pattern-matching algorithm. For example, Mootha et al.58 used a transcriptomic profile (derived from diabetic versus healthy muscle tissue) (Figure 2(a)) as the query, pathway-based gene sets (Figure 2 (e) and (d)) for the reference database, and GSEA for pattern-matching. On the other hand, Lamb et al.4 used gene signatures of HDAC inhibitors as the query, transcriptomic profiles as the reference database, and a modified version of GSEA. Although the workflow used by Mootha et al. is generally referred to as “pathway enrichment,” using the harmonized scheme presented here, we discuss how “enrichment” and “connectivity mapping” may be considered different types of similarity measures for comparing gene set objects comprised of gene signatures and transcriptomic profiles.
Figure 2.
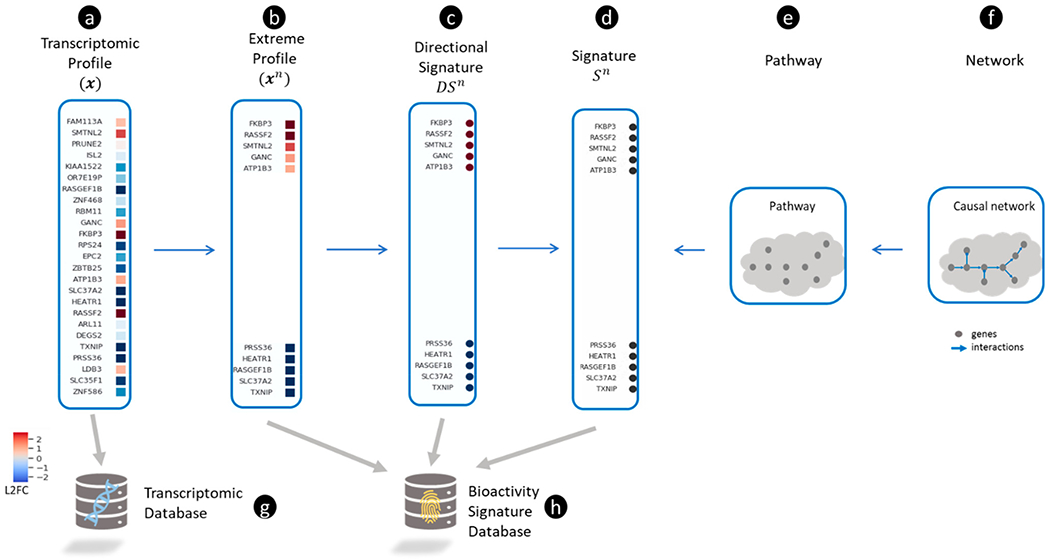
Representing transcriptomic profiles and gene signatures. (a) A transcriptomic profile
For example, the query object used by Lamb et al.5 was a gene signature derived from transcriptomic profiles of HDAC inhibitors in bladder and breast cancer cells.63 This gene signature for “HDAC inhibition” was defined by a set of up- and down-regulated genes (visualized in Figure 2(c)). In contrast, the reference objects were transcriptomic profiles (HYPERLINK Figure 2(a)) in the CMap v2 database. The similarity between the query gene signature for HDAC inhibition and each CMap v2 reference transcriptomic profile was measured using the same scoring metric as GSEA. Top-scoring matches with vorinostat and trichostatin A are both well-known HDAC inhibitors. In other words, GSEA “connected” the biological state of the query object, represented by a gene signature, with HDAC inhibition. This approach for connectivity mapping can be used in toxicity testing for new chemicals by generating gene signatures using transcriptomics, matching signatures with transcriptomic profiles for previously tested chemicals, and inferring putative connections with known chemicals’ mechanisms. Though connectivity mapping approaches measure the similarity between gene sets and transcriptomic profiles, there are subtle differences between them. First, Mootha et al. used a transcriptomic profile as the query object, whereas Lamb et al. used a gene signature. Second, Mootha et al. used pathway gene signatures, whereas Lamb et al. used transcriptomic profiles to define the reference database. Third, both approaches used slightly different similarity measures because they were comparing different types of objects. We believe that a harmonized scheme for encompassing the diverse array of published transcriptomic connectivity mapping approaches can be developed by formalizing the definition of query objects, reference objects, and similarity scoring measures.
Review outline
Although connectivity mapping can elucidate the mechanisms of action or toxicity of chemicals, the relative advantages of different approaches have not been discussed before. This review analyzes the gene set-based connectivity analysis pipeline in terms of reference database construction, gene signature generation, and similarity scoring measures. First, we provide a standardized terminology to describe the critical elements of gene set-based approaches to compare and highlight their unique contributions. Second, we introduce several transcriptomic and other data sources and outline the development of reference databases. Third, we discuss some of the main approaches for generating transcriptomic signatures. Fourth, we propose a detailed classification of connectivity scoring measures, including GSEA and other variants. Fifth, we describe approaches for evaluating confidence in results from connectivity mapping. Sixth, we consider different approaches for comparing the performance of different connectivity mapping algorithms. Finally, we discuss some of the opportunities and challenges of applying connectivity mapping approaches to toxicology. Our objective is to provide a conceptual overview of the entire connectivity mapping process at multiple levels, including high-level visual summaries, a formal terminology for comparing all connectivity mapping approaches, and detailed explanations of algorithms.
Key concepts and terminology
To compare the diversity of gene set-based connectivity mapping approaches, we define the key concepts and introduce terminology used in the remainder of this review. We begin with a description of different types of technologies routinely used for transcriptomic data generation. Next, we define a transcriptomic profile as the global differential gene expression data and discuss how gene signatures are created from transcriptomic profiles. Then we describe the different types of gene signatures that encompass transcriptomic profiles and gene sets that define canonical pathways. Lastly, we introduce key concepts about the relationships between gene signatures and transcriptomic profiles, and then use them to formalize similarity scoring measures. A visual overview of the different terms and their definitions are provided in Figure 1 and Table 1, respectively.
Table 1.
Glossary of transcriptomic terminology presented in this review.
| Transcriptomics |
Transcriptomics is defined as the measurement of large-scale (“global”) gene ( |
| Transcriptomic technologies | Transcriptomic technologies use different approaches to measure global gene expression by quantifying individual mRNA molecules. Most technologies synthesize complementary DNA (cDNA) from mRNA and use complementary oligonucleotide probes to specifically detect cDNA by hybridization. Examples of transcriptomic technologies include: microarrays, L1000 and RNA-Seq. |
| Transcriptomic data | Transcriptomic data (or gene expression data) from different technologies is generated from biological samples under different experimental conditions including normal vs. diseased, control vs. treated, etc. Two frequent types of treatments are chemical and genetic perturbations involving the knock-out or over-expression of specific genes. Transcriptomic data can be represented by at least four main levels where the higher levels of data are derived from the lower levels: raw data specific to the assay technology (L0), unnormalized mRNA data derived from L0 data using assay-specific processing (L1), normalized mRNA data that captures the absolute levels of expression for genes and is comparable across the study (L2), differential expression data that captures the change in mRNA levels from the control (and may have associated statistical significance scores) that is comparable across studies (L3). |
| Transcriptomic profile | We define the transcriptomic profile ( |
| Extreme transcriptomic profile | We define the extreme transcriptomic profile ( |
| Directional gene signature | A directional gene signature ( |
| Gene signature | A gene signature ( |
| Gene set object | We define a gene set object ( |
Transcriptomic technologies and data
Transcriptomics measures global gene expression in a biological specimen, and the term was coined for RNA-Seq technology.73 Here we use transcriptomics to refer to any high-throughput gene expression technology including, but not limited to, Affymetrix microarrays (used for building the CMap databases), the L1000 platform (using in the Library of Integrated Network-based Cellular Signatures (LINCS) database), and RNA-Seq technology. Transcriptomic data from different technologies are generally represented at four different levels: (L0) level 0 raw data specific to the assay technology, (L1) level 1 unnormalized mRNA data derived from raw data using assay-specific processes, (L2) level 2 normalized mRNA data, and (L3) level 3 differential gene expression data obtained by analyzing mRNA data between cases and controls. Each technology has varying needs for RNA purification but may or may not require complementary DNA (cDNA) synthesis. Affymetrix high-density microarrays hybridize each mRNA in a sample with thousands of oligonucleotide probes. Each mRNA sequence is mapped to a set of probes (called a probeset) designed to optimize the sensitivity and specificity of measurements. Affymetrix microarray L0 data are images, called cell intensity files (CEL), which are processes using image analysis tools to produce L1 data. L1 data is normalized74 using one of the available approaches75,76 to estimate L2 data as logarithm (base 2) intensity value for each transcript. LINCS data are generated using the L1000 platform.77 The LINCS platform employs flow cytometry to measure the relative abundance of mRNAs that hybridize to Luminex beads tagged with fluorescent oligonucleotide probes (producing Luminex bead array data as L0 files). The L0 flow cytometry data are deconvoluted to obtain L1 data, which are quantile-normalized to get L2 data on landmark genes (978) and imputed transcripts (12,336). RNA-Seq uses high-throughput DNA-sequencing technology to directly read the cDNA in samples producing mRNA sequence fragments, known as “reads,” as raw FASTQ files (L0). Each read in L0 is aligned with the known sequences of gene products or the entire genome to generate L1 data (for a review of best practices, see Conesa et al.78). RNA-Seq technology continues to evolve rapidly, and several modifications have been proposed to avoid cDNA synthesis (by using RNA directly) and to target specific genes (instead of sequencing all genes), including NPSeq,79 RASL-Seq,80 DRUG-Seq,81 and TempO-Seq.70 Such targeted RNA-Seq approaches can be cost-effective as they use oligonucleotide templates (also called probes) derived from specific regions in individual genes to measure their expression to produce L0 and L1 data. Validation studies have demonstrated the concordance between L1000 and microarrays,82 and targeted RNA-Seq, RNA-Seq, and microarrays83 for evaluating reference chemicals. The choice of transcriptomic platform comes down to reproducibility and cost. Therefore, new transcriptomic technologies that promise to lower the cost and efficiency of transcriptomic data generation continue to be developed (for example, see NanoString84).
Differential gene expression analysis
The L2 normalized mRNA data for each gene in the “cases” are compared with “controls” to calculate each gene’s differential expression. There are many statistical approaches for estimating confidence in each gene’s differential expression based on distributional intensity and count data assumptions.85–87 Batch-correction approaches can also improve differential expression estimates.88–90 The differential expression values for genes are reported as the ratio (or difference for log-transformed L2 data) between the cases (e.g., chemical treatments or diseased subjects) and the controls (e.g., untreated samples or normal subjects). Differential gene expression is generally reported in log2 fold-change (L2FC) units. When batch effects are significant, Z-scores offer another approach to estimating treatment effects. They can be averaged over batches and reported as moderated Z-scores.77 L2FC values or Z-scores have the following interpretation: positive/negative values mean that a gene is up-/down-regulated in a case versus control. The set of differential expression values for all genes defines the L3 transcriptomics data.
Transcriptomic profile
We define L3 data as the differential transcriptomic profile (
Extreme transcriptomic profile
Highly differentially expressed genes may be more informative than less differentially expressed ones. The extreme transcriptomic profile (
Gene signatures
A gene signature is a list of genes (also known as a “gene set”) whose collective activity represents a fingerprint of a biological state (mechanism, pathway, disease, etc.). We discuss two main types of gene signatures in this review. First, we define a “directional” gene signature (
Harmonizing gene signatures and transcriptomic profiles
Thus far, we have discussed four different concepts for representing biological states using gene expression: two types of transcriptomic profiles ({
Transcriptomic similarity measures
We can now define a similarity measure (
Table 2.
Summary of connectivity mapping methods
| Type | Method | Query | Ref | Publications |
|---|---|---|---|---|
| Aggregation-based |
eXtreme Sum (XS) |
|
|
Cheng et al. 2014 |
| eXtreme Mean |
|
|
||
| T-statistic (TS) |
|
|
Tian et al. 2005; Goeman et al. 2004, 2005 | |
| Ranksum statistic (RS) |
|
|
Barry, Nobel, and Wright 2005; Gower, Spira, and Lenburg 2011 | |
| GSEAa |
|
|
Mootha et al. 2003 | |
| GSEAb |
|
|
Subramanian et al. 2005 | |
| GSEAc |
|
|
Subramanian et al. 2007 | |
| Total enrichment score (TES) |
|
|
Iorio, Tagliaferri, and Bernardo 2009 | |
| Vector-based |
Pearson correlation |
|
|
Tenenbaum et al. 2008 |
| Spearman Correlation |
|
|
Tanner and Agarwal 2008; Zhang et al. 2009 | |
| Cosine |
|
|
Cheng et al. 2012 | |
| Jaccard index (JI) |
|
|
||
| Signed Jaccard (SJI) |
|
|
Zichen Wang et al. 2016 |
Reference databases
We consider two main types of reference databases (
Figure 4.
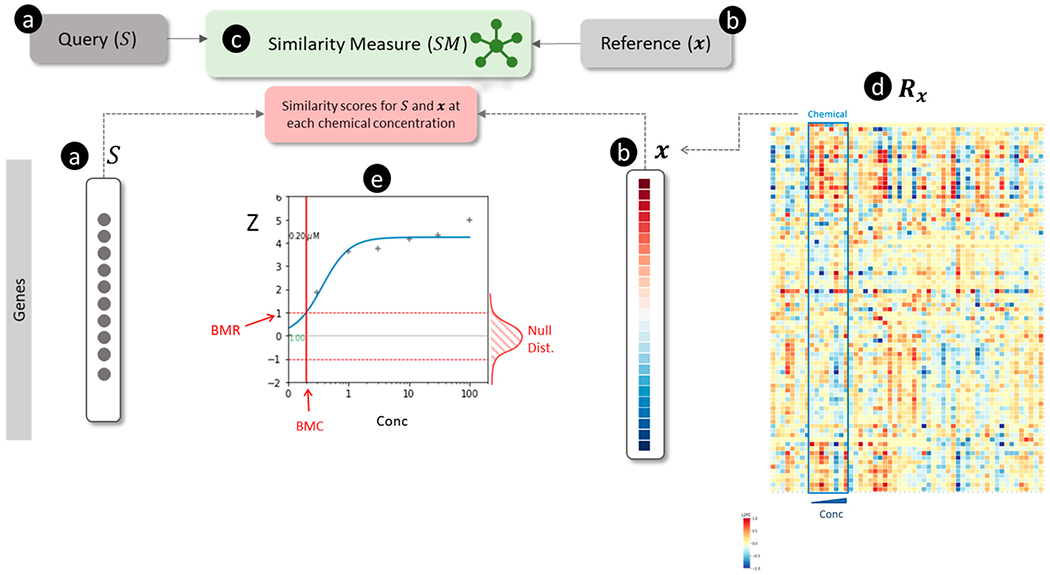
Overview of connectivity mapping for estimating chemical concentration-dependent scores for a signature. (a) The query is a non-directional gene signature (
Connectivity Map (CMap)
CMap v1 was initially developed to find relationships between chemicals, genes, and diseases.4 This dataset was produced by treating MCF7 (breast cancer), HL60 (leukemia), and SKMEL5 (melanoma) cell lines with 164 diverse chemicals at a 10 μM concentration for both 6 and 12 h. Transcriptional profiles were generated using Affymetrix GeneChip HGU133 measuring the levels of ~ 22,000 transcripts. In all, there were 564 unique transcriptomic profiles and 453 differential expression profiles (after comparing treatments and controls). Following the success of this approach, the same group produced CMap v2,65 in which three cell lines (MCF7, PC3, and HL60) were treated with 1,309 chemicals for 6 h to produce 6,100 differential expression profiles using the Affymetrix U133A GeneChip96 containing 22,215 transcripts associated with 13,609 genes. The entire CMap v2 database contains 1,294 chemical differential expression profiles in MCF7 cells, 1,182 profiles in PC3 cells, and 1,078 profiles in HL60 cells and is available for download from The Broad Institute. The raw Affymetrix data are normalized and processed (as described earlier) to generate a set of transcriptomic profiles,
Library of Integrated Network-based Cellular Signatures (LINCS)
Following the success of the CMap v2 project, the U.S. National Institutes for Health (NIH) funded the LINCS Consortium to expand the reference transcriptome database to study genetic (single gene over-expression or knockdown) and chemical perturbations producing a database containing more than 1,000,000 profiles.77 To achieve this 1000-fold scale-up of CMap v2, the LINCS Consortium developed computational methods to analyze a large compendium of expression data (12,031 Affymetrix gene expression profiles from GEO) to identify a subset of genes that could predict the entire transcriptome. Their analysis showed that using just 978 “landmark” transcripts could predict the expression of 82% of all genes. The L1000 platform measures these 978 genes (or 1,058 probes) using Luminex bead-based technology. It is, therefore, possible to infer the expression levels of 12,336 genes from the landmark 978 genes, and the resulting transcriptomic profiles are available as moderated Z-scores. The LINCS project has produced 1,319,138 L1000 profiles for 19,811 chemicals and 7,494 genetic perturbations. The L0, L1, L2, and L3 LINCS data are available from GEO as dataset GSE92742 and can also be interactively (or programmatically) analyzed via a cloud-based system (http://clue.io).
Gene Expression Omnibus
The US National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO)11 database is a public domain repository of author-submitted transcriptomics data that conforms to the MIAME (minimum information about a microarray experiment) standard.97 Unlike CMap and LINCS, GEO is a repository of published experimental studies and transcriptomics platforms. In GEO, highly multiplexed transcriptomics assays (like the Affymetrix HGU133 GeneChip and The Broad Institute’s L1000 array) are “platforms,” individual transcriptomics profiles are “samples,” and large-scale experiments are stored as a “series” of samples (like CMap v1, v2, and LINCS). A platform contains a set of probes directly linked to genes. The transcriptomic profile for a sample measures the levels for all probes. After substantial curation, a collection of biologically and statistically comparable samples in a series can be made available as a GEO DataSet. As of September 2021, GEO contains 4,348 DataSets, 160,597 series records, from 22,587 platforms and including 4,628,210 samples. All data in GEO can be freely downloaded and analyzed using the R programming language98 via the GEOquery99 package or using the Python programming language using the BioPython100 package.
ArrayExpress
The European Bioinformatics Institute (EBI) ArrayExpress12,101,102 database is the European counterpart to GEO, which also stores transcriptomics data in a MIAME-compliant format provided by authors in support of publications. In addition to transcriptomic data, ArrayExpress also maintains data from other molecule profiling technologies that include measurements of small molecules (metabolomics) and proteins (proteomics). Because metadata for transcriptomic profiles, including experimental model and treatment-related factors, are annotated using a controlled vocabulary,103 ArrayExpress can be more suitable for building automated computational workflows. Although initially intended as an integrated resource for all gene expression studies from GEO and the DNA Data Bank of Japan,104 the rapid growth of transcriptomics data has made this challenging. The lack of a central “index” of all transcriptomic studies makes it necessary to query ArrayExpress and GEO separately; however, some efforts have been undertaken to address this issue.105 Data in ArrayExpress can be searched and retrieved using the R ArrayExpress package106 and the Python BioServices package.107
Directional gene sets
With the availability of thousands of transcriptomics profiles in CMap, LINCS, GEO, and ArrayExpress,102 it is also feasible to automatically generate gene signatures for a rich range of biological contexts. Gene signatures have been automatically generated for many GEO datasets using detailed annotations of treatments.108–111 The specific genes included in a signature depend on the annotation of control (normal) and case (perturbed) samples in a study. Automated sample class interpretation (i.e., normal vs. perturbed) can be error-prone due to inconsistent annotations. Therefore, it is essential to manually-curate sample annotations in large public transcriptomic databases to generate valid gene signatures, which is a resource-intensive task. A crowd-sourcing approach has been recently used to develop CRowd Extracted Expression of Differential Signatures (CREEDS),112 which contains
Pathway and other gene sets
Pathway gene set databases can be constructed from canonical pathways that capture expert knowledge-based descriptions of biological processes. This has also been done comprehensively in MSigDB,113,114 which includes several canonical databases such as Reactome,115 Kyoto Encyclopedia for Genes and Genomes (KEGG),116 the National Cancer Institute (NCI) Pathway database117 and Gene Ontology.118 MSigDB also contains gene sets related to genetic and chemical perturbations, gene co-expression modules, transcription factor targets, etc., which are represented by
Connectivity scoring approaches
As stated earlier, gene set-based scoring can be stated generally as
Aggregation-based enrichment scoring approaches
Matching transcriptomic profiles to pathway signatures:
These SMa match transcriptomic profiles of samples against reference pathway databases (Illustrated in Figure 3). They calculate differences in the distributions of differential expression values in
Figure 3.
Overview of connectivity mapping as a pattern matching between (a) transcriptomic profile query and (b) non-directional signature reference using a (c) similarity measure showing illustrative examples of matches (f, g, h, and i). (a) The query is a transcriptomic profile
Different
There are three main versions of GSEA, and they are all based on a non-parametric aggregation approach. We discuss the first two versions of GSEA here as they are both of type
Connectivity scoring: 𝑆 𝑀 𝑎 ( 𝐷 𝑆 𝑞 , 𝒙 𝑟 )
The development of CMap required a new type of similarity measure for matching directional signatures (
Vector-based similarity scoring approaches 𝑆 𝑀 𝑣 ( 𝒙 𝑞 , 𝒙 𝑟 )
Vector-based approaches use different similarity measures to calculate
The first application of cosine similarity was based on using “extreme” transcriptomic profiles (described earlier), and the corresponding similarity measure was referred to as the extreme cosine score (XCos).137 Instead of cosine similarity, the correlation has also been used by several groups to calculate connectivity. Geneva120 uses Pearson and Spearman correlation coefficients between
The Jaccard index139 (or Jaccard similarity) is an even more straightforward approach for calculating the distance between binary representations of
Estimating significance of transcriptomic similarity scores
Evaluating confidence in similarity scores is important for determining the biological relevance of matches between
Instead of permutation-testing, it is also possible to derive
Evaluating transcriptomic similarity matching approaches
Given the breadth of techniques involved in connectivity mapping, one approach for evaluating their utility is to compare their performance using objective criteria. For most new connectivity mapping methods, this means evaluating performance by scoring hits between gene set objects from reference data sets using one of the baseline approaches (which are generally GSEAb and GSEAc). Alternatively, the performance of connectivity mapping can also be framed as a classification problem. For example, Mootha et al.58 analyzed differentially expressed genes from diabetic muscle samples and “classified” them as relevant for oxidative phosphorylation. This approach can be generalized for evaluating connectivity mapping; however, it requires an annotated set of positive and negative examples in reference databases.
One of the challenges in evaluating connectivity mapping approaches is that there are no gold-standard data sets of chemicals and targets. For example, the Anatomical Therapeutic Chemical (ATC) classification142 organizes drugs hierarchically based on four levels: the organ system at the top level, then therapeutic characteristics, and then the specific mechanism at level 4. One approach for classifying mechanisms by connectivity mapping developed by Iorio et al.93 used TES to identify similar drugs in CMap v2. They constructed a “drug network” based on connectivity scores and evaluated significance based on random pairs in CMap v2. Next, they determined whether similarity in the network predicted similarity in drug mechanisms labeled by ATC codes. After evaluating performance using the area under the receiver operating characteristic (AUROC) curve, they showed that close neighbors in the network shared mechanisms. While this analysis did not objectively compare different
The first evaluation of connectivity mapping approaches for predicting drug mechanisms was based on CMap v2 as the reference database with drug mechanisms labeled using ATC codes.72 This work compared mean-centering and differential expression analysis (based on treated and control samples) for creating the transcriptomic reference database, a range of signature sizes, different connectivity scoring methods (TES, GSEA, XCos), and used AUROC curves to compare the performance of these approaches for classifying ATC level 4 codes. Instead of using the area under the entire ROC curve, Cheng et al. measured the partial area under the curve (AUC) for a false positive rate of less than 0.1 (AUC0.1). Based on AUC0.1, the XCos vector similarity method outperformed KS and TES using gene signatures comprised of 100 up- and 100 down-regulated genes.
Transcriptomic Connectivity Mapping in Toxicology
Determining the potency and potential of chemicals for activating molecular targets that can lead to adverse health outcomes is a key challenge in toxicology.66 Though high-throughput screening (HTS) formats such as ToxCast 143,144 are more cost-effective than animal testing; there are far too many chemicals in commerce to evaluate using multiple HTS assays. High-throughput approaches such as RNA-Seq,9 and, more recently, TempO-Seq,70 are extremely promising and cost-effective for generating transcriptomic profiles for tens of thousands of chemical treatments. Connectivity mapping can be used to evaluate the potential targets and the off-target effects of a new drug or a chemical. Whether considering new chemical entities for drug discovery or untested environmental chemicals for public health protection, transcriptomic connectivity mapping is a robust and high-throughput alternative to the existing techniques.16 With a deadline of eliminating the use of mammalian test results by 2035,145 developing new approach methodologies (NAMs), which could efficiently provide information about chemical hazards and risks without using whole animals,146 is imperative for protecting public health and the environment. Connectivity mapping using transcriptomic data is a NAM-based methodology that will aid in realizing this vision.
Connectivity mapping has been used to characterize ecotoxicological chemical stressors using fish transcriptomic data.147 The authors constructed a reference database for 55 treatment conditions using transcriptomics data from fathead minnow and zebrafish (using a variety of gene expression platforms). Chemicals in the reference database were annotated with mechanisms using molecular initiating events (MIEs) in adverse outcome pathways (AOPs). Then they used sscMap133 to find connections between gene signatures derived from new samples and profiles in the reference database. De Abrew et al. investigated the mode of action (MOA) for 34 different chemicals using transcriptomic profiles measured in MCF7, Ishikawa, HepaRG, and HepG2 cells by comparing them with the CMap v2 database by similarity using hierarchical clustering and identified biologically-relevant connections.48 More recently, we have used multiple connectivity mapping methods presented in this review to solve three problems. First, we evaluated the reproducibility of transcriptional effects for reference chemicals in primary rat hepatocytes using TempO-Seq and Affymetrix data from OpenTG Gates.69 We found that Jaccard and cosine similarity was more accurate than GSEAc for correctly matching extreme transcriptomic profiles of chemicals produced by TempO-Seq and Affymetrix technologies. Second, we successfully used GSEAc to calculate the concentration-dependent effects of chemicals on pathways and directional signatures68 using an approach illustrated in Figure 4. Third, we developed consensus signatures of stress response pathways and used GSEAc to match them with transcriptomic profiles for reference perturbagens.148 These studies suggest the feasibility of applying connectivity mapping to evaluate environmental chemical toxicities using transcriptomics data.
Using connectivity mapping to evaluate the potential off-target effects of drugs or effects of environmental chemicals requires a computational pipeline with six components. First, a database containing gene signatures corresponding to the transcriptional effects of reference chemicals and canonical pathways. Large-scale transcriptomic data sets such as CMap,4 LINCS,95 and GEO11 can be used to create a reference gene signature (
The choice of similarity measure is essential in applying connectivity mapping to toxicology. Systematically comparing GSEA with other vector and aggregation-based approaches using benchmark data suggests that it has either similar or lower performance.119,120,123,126,127 Our initial findings also suggest that vector-based approaches combined with extreme transcriptomic profiles match chemicals to their known molecular targets more accurately than GSEA.69,151 On the other hand, aggregation-based approaches could be more suitable for concentration-response modeling (see Figure 4) to estimate biological pathway activating concentrations.68 Given the many steps involved in connectivity mapping workflows, further research on the contribution of different factors, including the choice of similarity measures, is necessary.
Discussion
The reproducibility,152 scalability,9,70 and broad biological coverage of transcriptomics make it feasible to profile millions of biological samples for chemical treatments, genetic manipulations, and diseases.5,11,95 In toxicology, connectivity mapping is being used to identify the putative targets of new chemicals,48,69,147 to determine their impact on ecological and human health via adverse outcome pathways,153 to demonstrate the robustness and reproducibility of transcriptional effects across different studies and technology platforms,69 and to estimate biological pathway activating concentrations.68 This makes transcriptomic data-driven connectivity mapping a powerful tool for screening the many thousands of chemicals in commerce154 for putative effects and potency estimates for a broad array of cellular pathways.
One of the potential novel applications of connectivity mapping is to provide potential biological analogues47,48 when a new chemical lacks structurally similar substances. In such cases, using transcriptomic profiles in place of structure descriptors can rapidly identify possible mechanisms, and other properties, for untested chemicals using reference databases.69
This review provides a systematic analysis of the key elements of connectivity mapping and a detailed tutorial to build workflows for evaluating chemical-induced toxicity using transcriptomic data. First, we proposed a coherent terminology to formalize diverse transcriptomic technologies (microarrays, L1000 and RNA-Seq). This terminology forms the basis of a uniform framework to represent transcriptomic profiles and produce gene signatures from them. Second, we used our proposed terminology to discuss some of the most widely used data sources for transcriptomic profiles (CMap, GEO, and LINCS) and gene signatures (MSigDB and CREEDS). Third, we formalized the connectivity mapping workflow in terms of a database search and retrieval task in which a query object is searched against a reference database using similarity measures to identify “hits.” Fourth, we classified published connectivity mapping approaches into two broad categories of similarity measures: vector-based and aggregation-based approaches. While vector-based approaches resemble similarity metrics used in other domains (e.g., cheminformatics), aggregation-based approaches are a new class of algorithms for measuring the similarity between transcriptomic profiles and gene signatures. Fifth, we reviewed the performance of aggregation and vector approaches reported in the literature on benchmark transcriptomic data sets. Beyond serving as a review of connectivity mapping approaches, we believe this manuscript can serve as a practical guide for implementing workflows by integrating public domain data and computational tools.
There are many limitations of connectivity mapping methods that should be addressed by future research to improve the utility of these approaches to toxicology and related disciplines. We summarize these into four broad categories: defining signatures of biological states, curating new benchmark data sets for evaluating connectivity mapping approaches, devising novel sensitive and specific similarity measures, and developing computational workflows that standardize and enable reproducible workflows. New strategies for developing reliable gene signatures will be vital for successfully applying connectivity mapping to interpret biological effects from transcriptomic profiles. First, there is considerable redundancy in published signatures (e.g., MSigDB113), which can produce multiple hits when searching transcriptomic profiles. Searching transcriptomic profiles against redundant signatures produces related hits that can hide the subtle but more biologically relevant effects. For example, there are dozens of signatures in MSigDB associated with DNA damage response, oxidative stress response, unfolded protein response, and other cellular stress response pathways. One approach for reducing redundancy is to aggregate related signatures into “consensus” signatures.155 Indeed, the idea of developing such consensus signatures is embodied in the Hallmark Signature collection of MSigDB.114 Second, published signatures may not wholly encompass the full range of phenomena observed in transcriptomic data. Using transcriptomic profiles for more extensive collections of chemical and genetic perturbations (e.g., from CMap, LINCS, etc.), it may be possible to fingerprint a more comprehensive array of chemical mechanisms or putative biomarkers of toxicity156 as gene signatures. Early transcriptomic technologies were limited by data reproducibility,157 which reduced their utility in finding reliable biomarkers. Although dealing with biological variability is still a challenge,158 technological advancements are improving data quality,68,69,152 making it feasible to transform transcriptomic profiles for specific perturbagens into gene signatures that can be used for connectivity mapping more reliably (e.g., shown visually in Figure 2). Therefore, additional research is necessary for combining profiles for a chemical (e.g., for different concentrations and time points) or for chemicals with similar mechanisms to develop new gene signatures. ML techniques could aid the development of such signatures when there are a sufficient number of profiles,159 simple statistical approaches may be adequate if there are just a handful of profiles to form such signatures.155 The problem is that similarity-based methods, such as connectivity mapping, require a sufficient number of relevant descriptors but are notoriously sensitive to irrelevant descriptors.23 The number of genes necessary for producing an accurate signature for identifying a specific mechanism from transcriptomic data can only be defined by empirical evaluation. For example, Lee et al. compared chemical-induced gene signatures (from primary rat hepatocytes using TempO-Seq data) of increasing sizes using different connectivity scoring approaches with a legacy data set (Affymetrix data from Open TG-GATES160) with varying results by chemical, mechanism, and treatment concentration.69 As expected, 8 and 200 μM of WY14643, which is a peroxisome-proliferator activated receptor alpha (PPARα) agonist, produced hits with other PPARα activators in Open TG-GATES. Similarly, valproic acid (400 and 10,000 μM) produced hits with other PPARα activators in Open TG-GATES using different vector-based approaches. On the other hand, even a 1000 μM acetaminophen treatment did not find any relevant matches. While using concentration-response data could avoid false negatives, optimizing the choice and number of genes included in signatures plays an important role in finding mechanisms of chemicals with subtle transcriptional effects.
The robustness of connectivity mapping approaches for differentiating actual biological signals from the noise requires further systematic evaluations using benchmark transcriptomics data sets derived from chemicals with known bioactivities studied in appropriate cell types using relevant concentrations and exposure durations. Although the ATC classification142 helps annotate drugs, this scheme is not ideal for annotating environmental chemicals. Other efforts are underway to integrate evidence about chemical bioactivity from disparate structured150 and unstructured151 sources to define potential reference chemicals for specific molecular and cellular targets. Once chemicals have been assigned to different classes of targets, then transcriptomic profiles for these chemicals can be retrieved from CMap, LINCS, or GEO to build gene signatures using the approaches discussed earlier. While connectivity mapping with these reference signatures can help assign putative targets or pathways to untested chemicals with high sensitivity, confident assessment of specificity is still challenging. Therefore, further work on improving specificity, by either finding additional perturbagens that do not activate a given target (i.e., negatives) or by using different randomization strategies to create “null” chemicals, is also important. Lastly, new software tools are needed for implementing different connectivity mapping workflows that can be tailored to specific problems and executed efficiently for large-scale data sets.
We have attempted to organize the spectrum of connectivity mapping approaches into vector- and aggregation-based approaches. Vector-based similarity measures are widely used in cheminformatics for finding chemical analogs using structure descriptors. Interestingly, vector approaches are also helpful in matching transcriptomic profiles and gene signatures. On the other hand, aggregation-based methods are unique to analyzing transcriptomic data, with GSEA as one of the most widely used techniques. Systematic comparisons of GSEA with other vector and aggregation-based approaches using benchmark data suggest that it may not always be the most accurate method.119,120,123,126,127 For toxicology applications, our findings to date suggest that vector-based approaches are more effective for identifying putative molecular targets,69,151 but aggregation-based approaches are more suitable for concentration-response modeling (see Figure 4) to estimate biological pathway activating concentrations.68 Further research into the relative merits of vector- and aggregation-based approaches for toxicology applications could address some of these questions.
Novel connectivity mapping approaches beyond vector- and aggregation-based techniques are under active development, and two promising research areas are worth mentioning. First, dimensionality reduction of transcriptomic profiles to find more biologically meaningful latent representations (e.g., for molecular targets) could overcome some challenges in finding optimal gene signatures from noisy data. For instance, probabilistic connectivity mapping (ProbCMap161) uses latent factor models, which aggregate information across genes using different statistical models to construct a low-dimension representation of transcriptomic profiles. ProbCMap uses group factor analysis,162 sparse factor analysis,163 and Bayesian principal components analysis to generate low-dimensional vector representations of transcriptomic profiles and measure similarity between them using Pearson correlation. More recently, deep learning methods164 based on multilayer artificial neural networks are enabling an exciting wave of novel data-driven approaches to elucidate latent representations of biological mechanisms from transcriptomic data 165 and even predict transcriptomic signatures based on transcription factor activity.166 Second, traditional gene signatures can be enhanced with computational approaches for analyzing genetic regulatory and signaling networks167–169 to predict the activation of transcription factors from transcriptomic data.170 Although we have not described these two approaches systematically in this review, the background and formal basis for analyzing connectivity mapping will help readers to place these in context. Our future work will expand on new connectivity mapping strategies based on deep learning and network analysis for identifying molecular targets of chemicals and drugs using transcriptomic data.
Conclusion
Connectivity mapping assumes that if two transcriptomic profiles are similar, it is due to a common biological state or process. If two chemicals produce similar transcriptomic profiles, then it could mean that they act via similar mechanisms. Therefore, connectivity mapping can infer the putative molecular targets of new chemicals based on existing chemicals or the toxicological properties of new chemicals based on known toxicants.16 Connectivity mapping can be used for finding biological analogues,48 for determining the mechanism of action,69,147 and estimating pathway activating concentrations for chemicals by coupling similarity scores with concentration-response modeling.68 This review provides the relevant background and in-depth explanation of connectivity mapping workflows to address toxicology problems. It also lays out a roadmap for future research to address current challenges. This work is a conceptual overview for those interested in learning about the utility of connectivity mapping in the context of NAMs, practitioners interested in using connectivity mapping pipelines in their workflows, and researchers interested in developing novel approaches that advance the state-of-the-art.
Acknowledgments
The authors thank Dan Villeneuve and John Cowden for their insightful comments on this manuscript.
Funding Information
The USEPA, through its Office of Research and Development, provided funding for this research. J.N. was supported by appointments to the Research Participation Program of the USEPA, Office of Research and Development, administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the USEPA.
Biographies
Author Biographies
Dr. Imran Shah is a computational systems biologist at the US EPA Center for Computational Toxicology and Exposure (CCTE), where he develops new data-driven methods to predict chemical effects and toxicological tipping points. His research aims to reduce dependence on animal testing using artificial Intelligence, machine learning, and multi-scale models trained with large-scale chemical and bioactivity data. He also translates these models into interactive tools to support chemical safety decisions. He completed his B.Sc. in physics from Imperial College and a Ph.D. in computational biology from George Mason University.
Dr. Joseph Bundy is a biologist at the US EPA CCTE. He has experience analyzing and integrating high-dimensional molecular data sets focusing on transcriptomics. His current work involves leveraging machine learning techniques to predict the activation of Molecular Initiating Events in biological systems perturbed by chemical exposure.
Dr. Bryant Chambers is a post-doctoral research fellow in the US EPA CCTE. He is a computational biologist with experience in high-throughput transcriptomic chemical hazard screening. His research focuses on the role of stress response networks in toxicological outcomes, adaptive antimicrobial resistance, and mechanisms of nanotoxicity.
Dr. Logan E. Everett is a bioinformatics scientist in the US EPA CCTE. His expertise includes genomics, statistics, genetics, and molecular biology. His research at EPA is focused on advancing the application of high-throughput transcriptomics in chemical safety screening.
Dr. Derik Haggard is a scientific analyst at the US EPA CCTE. He has expertise in computational toxicology, developing high-throughput transcriptomics analysis pipelines, and assisting in developing tools, applications, software, and databases used in the chemical hazard characterization and risk assessment process.
Dr. Joshua Harrill is a cellular and molecular toxicologist at the US EPA CCTE. His expertise includes in vitro toxicology, specifically the application of transcriptomics, high-content imaging and other complementary technologies for high-throughput chemical hazard screening, characterization, and informing chemical risk assessment.
Dr. Richard S. Judson is a bioinformatics scientist at the US EPA CCTE, where he is developing computer models and databases to predict toxicological effects of chemicals. His current research includes in vitro and in vivo database development and using these to build models to predict the behavior of new chemicals, and deriving pathway-based approaches using high-throughput transcriptomics data. He has published in computational biology and chemistry, bioinformatics, genomics, human genetics, toxicology, and applied mathematics. He has a BA in Chemistry and Chemical Physics from Rice University and an MA and PhD in Chemistry from Princeton University.
Dr. Johanna Nyffeler is a post-doctoral research fellow at the US EPA CCTE. She focuses on developing new approach methodologies (NAMs) for toxicity testing, including high-content imaging and cell painting. She won the Lush Prize (2020) for her innovative research on assessing the developmental neurotoxicity of environmental chemicals using cell painting in neurons. Dr. Nyffeler completed her BS in biochemistry from the University of Fribourg, MS in genetics from the University of Zurich, and PhD in toxicology from the University of Konstanz.
Dr. Grace Patlewicz is currently a research chemist at the US EPA CCTE. She started her career at Unilever UK, before moving to the EC Joint Research Centre in Italy and then to DuPont in the US. A chemist and toxicologist by training, her research interests have focused on developing and applying QSARs and read-across for regulatory purposes. She has authored over 150 journal publications and book chapters, chaired various industry groups, and contributed to developing technical guidance for QSARs and chemical categories under various OECD programs.
Footnotes
Competing financial interests: The authors declare they have no actual or potential competing financial interests
Disclaimer: The views expressed in this article are those of the authors and do not necessarily represent the views or policies of the U.S. Environmental Protection Agency.
Conflict of Interest
The authors declare no conflict of interest. This manuscript has been reviewed by the Center for Computational Toxicology and Exposure, Office of Research and Development, U.S. Environmental Protection Agency, and approved for publication. Approval does not signify that the contents reflect the views of the Agency, nor does mention of trade names or commercial products constitute endorsement or recommendation for use.
References
- (1).Harrill J; Shah I; Setzer RW; Haggard D; Auerbach S; Judson R; Thomas RS Considerations for Strategic Use of High-Throughput Transcriptomics Chemical Screening Data in Regulatory Decisions. Curr. Opin. Toxicol 2019, 15. 10.1016/j.cotox.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).McKenna NJ; O’Malley BW Combinatorial Control of Gene Expression by Nuclear Receptors and Coregulators. Cell 2002, 108 (4), 465–474. 10.1016/S0092-8674(02)00641-4. [DOI] [PubMed] [Google Scholar]
- (3).Simmons SO; Fan C-Y; Ramabhadran R Cellular Stress Response Pathway System as a Sentinel Ensemble in Toxicological Screening. Toxicol. Sci 2009, 111 (2), 202–225. 10.1093/toxsci/kfp140. [DOI] [PubMed] [Google Scholar]
- (4).Lamb J; Crawford ED; Peck D; Modell JW; Blat IC; Wrobel MJ; Lerner J; Brunet J-P; Subramanian A; Ross KN; Reich M; Hieronymus H; Wei G; Armstrong SA; Haggarty SJ; Clemons PA; Wei R; Carr SA; Lander ES; Golub TR The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease. Science 2006, 313 (5795), 1929–1935. 10.1126/science.1132939. [DOI] [PubMed] [Google Scholar]
- (5).Lamb J The Connectivity Map: A New Tool for Biomedical Research. Nat. Rev. Cancer 2007, 7 (1), 54–60. 10.1038/nrc2044. [DOI] [PubMed] [Google Scholar]
- (6).Lander ES; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409 (6822), 860–921. 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- (7).Schena M; Shalon D; Davis RW; Brown PO Quantitative Monitoring of Gene Expression Patterns with a Complementary DNA Microarray. Science 1995, 270 (5235), 467–470. 10.1126/SCIENCE.270.5235.467. [DOI] [PubMed] [Google Scholar]
- (8).Lipshutz RJ; Fodor SPA; Gingeras TR; Lockhart DJ High Density Synthetic Oligonucleotide Arrays. Nat. Genet 1999, 21 (1s), 20–24. 10.1038/4447. [DOI] [PubMed] [Google Scholar]
- (9).Wang Z; Gerstein M; Snyder M RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet 2009, 10 (1), 57–63. 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Harrill J; Shah I; Setzer RW; Haggard D; Auerbach S; Judson R; Thomas RS Considerations for Strategic Use of High-Throughput Transcriptomics Chemical Screening Data in Regulatory Decisions. Curr. Opin. Toxicol 2019. 10.1016/J.COTOX.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Barrett T; Wilhite SE; Ledoux P; Evangelista C; Kim IF; Tomashevsky M; Marshall KA; Phillippy KH; Sherman PM; Holko M; Yefanov A; Lee H; Zhang N; Robertson CL; Serova N; Davis S; Soboleva A NCBI GEO: Archive for Functional Genomics Data Sets - Update. Nucleic Acids Res. 2013, 41 (D1), 991–995. 10.1093/nar/gks1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Athar A; Füllgrabe A; George N; Iqbal H; Huerta L; Ali A; Snow C; Fonseca NA; Petryszak R; Papatheodorou I; Sarkans U; Brazma A ArrayExpress Update - From Bulk to Single-Cell Expression Data. Nucleic Acids Res. 2019, 47 (D1), D711–D715. 10.1093/nar/gky964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Keenan AB; Jenkins SL; Jagodnik KM; Koplev S; He E; Torre D; Wang Z; Dohlman AB; Silverstein MC; Lachmann A; Kuleshov MV; Ma’ayan A; Stathias V; Terryn R; Cooper D; Forlin M; Koleti A; Vidovic D; Chung C; Schürer SC; Vasiliauskas J; Pilarczyk M; Shamsaei B; Fazel M; Ren Y; Niu W; Clark NA; White S; Mahi N; Zhang L; Kouril M; Reichard JF; Sivaganesan S; Medvedovic M; Meller J; Koch RJ; Birtwistle MR; Iyengar R; Sobie EA; Azeloglu EU; Kaye J; Osterloh J; Haston K; Kalra J; Finkbiener S; Li J; Milani P; Adam M; Escalante-Chong R; Sachs K; Lenail A; Ramamoorthy D; Fraenkel E; Daigle G; Hussain U; Coye A; Rothstein J; Sareen D; Ornelas L; Banuelos M; Mandefro B; Ho R; Svendsen CN; Lim RG; Stocksdale J; Casale MS; Thompson TG; Wu J; Thompson LM; Dardov V; Venkatraman V; Matlock A; Van Eyk JE; Jaffe JD; Papanastasiou M; Subramanian A; Golub TR; Erickson SD; Fallahi-Sichani M; Hafner M; Gray NS; Lin J-R; Mills CE; Muhlich JL; Niepel M; Shamu CE; Williams EH; Wrobel D; Sorger PK; Heiser LM; Gray JW; Korkola JE; Mills GB; LaBarge M; Feiler HS; Dane MA; Bucher E; Nederlof M; Sudar D; Gross S; Kilburn DF; Smith R; Devlin K; Margolis R; Derr L; Lee A; Pillai A The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6 (1), 13–24. 10.1016/J.CELS.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Qu XA; Rajpal DK Applications of Connectivity Map in Drug Discovery and Development. Drug Discovery Today. December 2012, pp 1289–1298. 10.1016/j.drudis.2012.07.017. [DOI] [PubMed] [Google Scholar]
- (15).Iorio F; Rittman T; Ge H; Menden M; Saez-Rodriguez J Transcriptional Data: A New Gateway to Drug Repositioning? Drug Discov. Today 2013, 18 (7–8), 350–357. 10.1016/j.drudis.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Smalley JL; Gant TW; Zhang S-D Application of Connectivity Mapping in Predictive Toxicology Based on Gene-Expression Similarity. Toxicology 2010, 268 (3), 143–146. 10.1016/J.TOX.2009.09.014. [DOI] [PubMed] [Google Scholar]
- (17).Gioia D; Bertazzo M; Recanatini M; Masetti M; Cavalli A Dynamic Docking: A Paradigm Shift in Computational Drug Discovery. Molecules 2017, 22 (11), 2029. 10.3390/molecules22112029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Rachman MM; Barril X; Hubbard RE Predicting How Drug Molecules Bind to Their Protein Targets. Curr. Opin. Pharmacol 2018, 42, 34–39. 10.1016/j.coph.2018.07.001. [DOI] [PubMed] [Google Scholar]
- (19).Colwell LJ Statistical and Machine Learning Approaches to Predicting Protein-Ligand Interactions. Curr. Opin. Struct. Biol 2018, 49, 123–128. 10.1016/j.sbi.2018.01.006. [DOI] [PubMed] [Google Scholar]
- (20).Lionta E; Spyrou G; Vassilatis DK; Cournia Z Structure-Based Virtual Screening for Drug Discovery: Principles, Applications and Recent Advances. Curr. Top. Med. Chem 2014, 14 (16), 1923–1938. 10.2174/1568026614666140929124445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jumper J; Evans R; Pritzel A; Green T; Figurnov M; Ronneberger O; Tunyasuvunakool K; Bates R; Žídek A; Potapenko A; Bridgland A; Meyer C; Kohl SAA; Ballard AJ; Cowie A; Romera-Paredes B; Nikolov S; Jain R; Adler J; Back T; Petersen S; Reiman D; Clancy E; Zielinski M; Steinegger M; Pacholska M; Berghammer T; Bodenstein S; Silver D; Vinyals O; Senior AW; Kavukcuoglu K; Kohli P; Hassabis D Highly Accurate Protein Structure Prediction with AlphaFold. Nat. 2021 5967873 2021, 596 (7873), 583–589. 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Mullard A What Does AlphaFold Mean for Drug Discovery? Nat. Rev. Drug Discov 2021. 10.1038/D41573-021-00161-0. [DOI] [PubMed] [Google Scholar]
- (23).Aha DW; Kibler D; Albert MK Instance-Based Learning Algorithms. Mach. Learn 1991, 6 (1), 37–66. 10.1007/BF00153759. [DOI] [Google Scholar]
- (24).Rogers D; Hahn M Extended-Connectivity Fingerprints. J. Chem. Inf. Model 2010, 50 (5), 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- (25).Bero SA; Muda AK; Choo YH; Muda NA; Pratama SF Similarity Measure for Molecular Structure: A Brief Review. J. Phys. Conf. Ser 2017, 892 (1), 012015. 10.1088/1742-6596/892/1/012015. [DOI] [Google Scholar]
- (26).van Laarhoven T; Marchiori E Predicting Drug-Target Interactions for New Drug Compounds Using a Weighted Nearest Neighbor Profile. PLoS One 2013, 8 (6), e66952. 10.1371/journal.pone.0066952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Stumpfe D; Bajorath J Exploring Activity Cliffs in Medicinal Chemistry. J. Med. Chem 2012, 55 (7), 2932–2942. 10.1021/JM201706B. [DOI] [PubMed] [Google Scholar]
- (28).Patlewicz G; Helman G; Pradeep P; Shah I Navigating through the Minefield of Read-across Tools: A Review of in Silico Tools for Grouping. Comput. Toxicol 2017, 3. 10.1016/j.comtox.2017.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shah I; Liu J; Judson RS; Thomas RS; Patlewicz G Systematically Evaluating Read-across Prediction and Performance Using a Local Validity Approach Characterized by Chemical Structure and Bioactivity Information. Regul. Toxicol. Pharmacol 2016, 79, 12–24. 10.1016/j.yrtph.2016.05.008. [DOI] [PubMed] [Google Scholar]
- (30).Helman G; Patlewicz G; Shah I Quantitative Prediction of Repeat Dose Toxicity Values Using GenRA. Regul. Toxicol. Pharmacol 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Helman G; Shah I; Patlewicz G Transitioning the Generalised Read-across Approach (GenRA) to Quantitative Predictions: A Case Study Using Acute Oral Toxicity Data. Comput. Toxicol 2019, 12, 100097. 10.1016/J.COMTOX.2019.100097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Tate T; Wambaugh J; Patlewicz G; Shah I Repeat-Dose Toxicity Prediction with Generalized Read-Across (GenRA) Using Targeted Transcriptomic Data: A Proof-of-Concept Case Study. Comput. Toxicol 2021, 19, 100171. 10.1016/j.comtox.2021.100171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Judson R; Elloumi F; Woodrow RW; Li Z; Shah I A Comparison of Machine Learning Algorithms for Chemical Toxicity Classification Using a Simulated Multi-Scale Data Model. BMC Bioinformatics 2008, 9. 10.1186/1471-2105-9-241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Muratov EN; Bajorath J; Sheridan RP; Tetko IV; Filimonov D; Poroikov V; Oprea TI; Baskin II; Varnek A; Roitberg A; Isayev O; Curtalolo S; Fourches D; Cohen Y; Aspuru-Guzik A; Winkler DA; Agrafiotis D; Cherkasov A; Tropsha A QSAR without Borders. Chem. Soc. Rev 2020, 49 (11), 3525–3564. 10.1039/d0cs00098a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Mansouri K; Abdelaziz A; Rybacka A; Roncaglioni A; Tropsha A; Varnek A; Zakharov A; Worth A; Richard AM; Grulke CM; Trisciuzzi D; Fourches D; Horvath D; Benfenati E; Muratov E; Wedebye EB; Grisoni F; Mangiatordi GF; Incisivo GM; Hong H; Ng HW; Tetko IV; Balabin I; Kancherla J; Shen J; Burton J; Nicklaus M; Cassotti M; Nikolov NG; Nicolotti O; Andersson PL; Zang Q; Politi R; Beger RD; Todeschini R; Huang R; Farag S; Rosenberg SA; Slavov S; Hu X; Judson RS CERAPP: Collaborative Estrogen Receptor Activity Prediction Project. Environ. Health Perspect 2016, 124 (7), 1023–1033. 10.1289/ehp.1510267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mansouri K; Kleinstreuer N; Abdelaziz AM; Alberga D; Alves VM; Andersson PL; Andrade CH; Bai F; Balabin I; Ballabio D; Benfenati E; Bhhatarai B; Boyer S; Chen J; Consonni V; Farag S; Fourches D; García-Sosa AT; Gramatica P; Grisoni F; Grulke CM; Hong H; Horvath D; Hu X; Huang R; Jeliazkova N; Li J; Li X; Liu H; Manganelli S; Mangiatordi GF; Maran U; Marcou G; Martin T; Muratov E; Nguyen DT; Nicolotti O; Nikolov NG; Norinder U; Papa E; Petitjean M; Piir G; Pogodin P; Poroikov V; Qiao X; Richard AM; Roncaglioni A; Ruiz P; Rupakheti C; Sakkiah S; Sangion A; Schramm KW; Selvaraj C; Shah I; Sild S; Sun L; Taboureau O; Tang Y; Tetko IV; Todeschini R; Tong W; Trisciuzzi D; Tropsha A; Van Den Driessche G; Varnek A; Wang Z; Wedebye EB; Williams AJ; Xie H; Zakharov AV; Zheng Z; Judson RS Compara: Collaborative Modeling Project for Androgen Receptor Activity. Environ. Health Perspect 2020, 128 (2), 1–17. 10.1289/EHP5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Geiss KT; Frazier JM QSAR Modeling of Oxidative Stress in Vitro Following Hepatocyte Exposures to Halogenated Methanes. Toxicol. In Vitro 2001, 15 (4–5), 557–563. 10.1016/S0887-2333(01)00063-7. [DOI] [PubMed] [Google Scholar]
- (38).Capuzzi SJ; Politi R; Isayev O; Farag S; Tropsha A QSAR Modeling of Tox21 Challenge Stress Response and Nuclear Receptor Signaling Toxicity Assays. Front. Environ. Sci 2016, 0 (FEB), 3. 10.3389/FENVS.2016.00003. [DOI] [Google Scholar]
- (39).Myshkin E; Brennan R; Khasanova T; Sitnik T; Serebriyskaya T; Litvinova E; Guryanov A; Nikolsky Y; Nikolskaya T; Bureeva S Prediction of Organ Toxicity Endpoints by QSAR Modeling Based on Precise Chemical-Histopathology Annotations. Chem. Biol. Drug Des 2012, 80 (3), 406–416. 10.1111/j.1747-0285.2012.01411.x. [DOI] [PubMed] [Google Scholar]
- (40).Mulliner D; Schmidt F; Stolte M; Spirkl H-P; Czich A; Amberg A Computational Models for Human and Animal Hepatotoxicity with a Global Application Scope. Chem. Res. Toxicol 2016, 29 (5), 757–767. 10.1021/ACS.CHEMRESTOX.5B00465. [DOI] [PubMed] [Google Scholar]
- (41).Pabon NA; Xia Y; Estabrooks SK; Ye Z; Herbrand AK; Süß E; Biondi RM; Assimon VA; Gestwicki JE; Brodsky JL; Camacho CJ; Bar-Joseph Z Predicting Protein Targets for Drug-like Compounds Using Transcriptomics. PLOS Comput. Biol 2018, 14 (12), e1006651. 10.1371/JOURNAL.PCBI.1006651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Szalai B; Subramanian V; Holland CH; Alföldi R; Puskás LG; Saez-Rodriguez J Signatures of Cell Death and Proliferation in Perturbation Transcriptomics Data - from Confounding Factor to Effective Prediction. Nucleic Acids Res 2019, 47 (19), 10010–10026. 10.1093/nar/gkz805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Hizukuri Y; Sawada R; Yamanishi Y Predicting Target Proteins for Drug Candidate Compounds Based on Drug-Induced Gene Expression Data in a Chemical Structure-Independent Manner. BMC Med. Genomics 2015, 8 (1), 82. 10.1186/s12920-015-0158-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Xie L; He S; Song X; Bo X; Zhang Z Deep Learning-Based Transcriptome Data Classification for Drug-Target Interaction Prediction. BMC Genomics 2018 197 2018, 19 (7), 93–102. 10.1186/S12864-018-5031-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Rueda-Zárate HA; Imaz-Rosshandler I; Cárdenas-Ovando RA; Castillo-Fernández JE; Noguez-Monroy J; Rangel-Escareño C A Computational Toxicogenomics Approach Identifies a List of Highly Hepatotoxic Compounds from a Large Microarray Database. PLoS One 2017, 12 (4). 10.1371/JOURNAL.PONE.0176284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Sumsion GR; Bradshaw MS; Beales JT; Ford E; Caryotakis GRG; Garrett DJ; Lebaron ED; Nwosu IO; Piccolo SR Diverse Approaches to Predicting Drug-Induced Liver Injury Using Gene-Expression Profiles. Biol. Direct 2020, 15 (1). 10.1186/S13062-019-0257-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Low Y; Sedykh A; Fourches D; Golbraikh A; Whelan M; Rusyn I; Tropsha A Integrative Chemical–Biological Read-Across Approach for Chemical Hazard Classification. Chem. Res. Toxicol 2013, 26 (8), 1199–1208. 10.1021/tx400110f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).De Abrew KN; Kainkaryam RM; Shan YK; Overmann GJ; Settivari RS; Wang X; Xu J; Adams RL; Tiesman JP; Carney EW; Naciff JM; Daston GP Grouping 34 Chemicals Based on Mode of Action Using Connectivity Mapping. Toxicol. Sci 2016, 151 (2), 447–461. 10.1093/toxsci/kfw058. [DOI] [PubMed] [Google Scholar]
- (49).Stockwell BR Chemical Genetics: Ligand-Based Discovery of Gene Function. Nature Reviews Genetics. European Association for Cardio-Thoracic Surgery 2000, pp 116–125. 10.1038/35038557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Eisen MB; Spellman PT; Brown PO; Botstein D Cluster Analysis and Display of Genome-Wide Expression Patterns. Proc. Natl. Acad. Sci. U. S. A 1998, 95 (25), 14863–14868. 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).DeRisi JL; Iyer VR; Brown PO Exploring the Metabolic and Genetic Control of Gene Expression on a Genomic Scale. Science (80-. ). 1997, 278 (5338), 680–686. 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- (52).Hughes TR; Marton MJ; Jones AR; Roberts CJ; Stoughton R; Armour CD; Bennett HA; Coffey E; Dai H; He YD; Kidd MJ; King AM; Meyer MR; Slade D; Lum PY; Stepaniants SB; Shoemaker DD; Gachotte D; Chakraburtty K; Simon J; Bard M; Friend SH Functional Discovery via a Compendium of Expression Profiles. Cell 2000, 102 (1), 109–126. 10.1016/S0092-8674(00)00015-5. [DOI] [PubMed] [Google Scholar]
- (53).Merlos M; Romero L; Zamanillo D; Plata-Salamán C; Vela JM Sigma-1 Receptor and Pain. Handb. Exp. Pharmacol 2017, 244, 131–161. 10.1007/164_2017_9. [DOI] [PubMed] [Google Scholar]
- (54).Moebius FF; Reiter RJ; Hanner M; Glossmann H High Affinity of Sigma 1 -Binding Sites for Sterol Isomerization Inhibitors: Evidence for a Pharmacological Relationship with the Yeast Sterol C 8 -C 7 Isomerase. Br. J. Pharmacol 1997, 121 (1), 1–6. 10.1038/sj.bjp.0701079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Fisher RA On the Interpretation of X2 from Contingency Tables, and the Calculation of P. J. R. Stat. Soc 1922, 85 (1), 87. 10.2307/2340521. [DOI] [Google Scholar]
- (56).Khatri P; Draghici S Ontological Analysis of Gene Expression Data: Current Tools, Limitations, and Open Problems. Bioinformatics 2005, 21 (18), 3587–3595. 10.1093/bioinformatics/bti565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Rivals I; Personnaz L; Taing L; Potier M-C Enrichment or Depletion of a GO Category within a Class of Genes: Which Test? Bioinformatics 2007, 23 (4), 401–407. 10.1093/bioinformatics/btl633. [DOI] [PubMed] [Google Scholar]
- (58).Mootha VK; Lindgren CM; Eriksson K-F; Subramanian A; Sihag S; Lehar J; Puigserver P; Carlsson E; Ridderstråle M; Laurila E; Houstis N; Daly MJ; Patterson N; Mesirov JP; Golub TR; Tamayo P; Spiegelman B; Lander ES; Hirschhorn JN; Altshuler D; Groop LC PGC-1α-Responsive Genes Involved in Oxidative Phosphorylation Are Coordinately Downregulated in Human Diabetes. Nat. Genet 2003, 34 (3), 267–273. 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- (59).Subramanian A; Tamayo P; Mootha VK; Mukherjee S; Ebert BL; Gillette MA; Paulovich A; Pomeroy SL; Golub TR; Lander ES; Mesirov JP Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (43), 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Huang DW; Sherman BT; Lempicki RA Bioinformatics Enrichment Tools: Paths toward the Comprehensive Functional Analysis of Large Gene Lists. Nucleic Acids Res. 2009, 37 (1), 1–13. 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Khatri P; Sirota M; Butte AJ 10 Years of Pathway Analysis : Current Approaches and Outstanding Challenges - Text S2. 2012, 10–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Nam D; Kim S-Y Gene-Set Approach for Expression Pattern Analysis. Brief. Bioinform 2008, 9 (3), 189–197. 10.1093/bib/bbn001. [DOI] [PubMed] [Google Scholar]
- (63).Glaser KB; Li J; Aakre ME; Morgan DW; Sheppard G; Stewart KD; Pollock J; Lee P; O’Connor CZ; Anderson SN; Mussatto DJ; Wegner CW; Moses HL Transforming Growth Factor Beta Mimetics: Discovery of 7-[4-(4-Cyanophenyl)Phenoxy]-Heptanohydroxamic Acid, a Biaryl Hydroxamate Inhibitor of Histone Deacetylase. Mol. Cancer Ther 2002, 1 (10), 759–768. [PubMed] [Google Scholar]
- (64).López IP; Marti A; Milagro FI; Zulet M, de los A; Moreno-Aliaga MJ; Martinez JA; De Miguel C DNA Microarray Analysis of Genes Differentially Expressed in Diet-Induced (Cafeteria) Obese Rats. Obes. Res 2003, 11 (2), 188–194. 10.1038/oby.2003.30. [DOI] [PubMed] [Google Scholar]
- (65).Lamb J Connectivity Map v2. https://portals.broadinstitute.org/cmap/.
- (66).Browne P; Noyes PD; Casey WM; Dix DJ Application of Adverse Outcome Pathways to U.S. EPA’s Endocrine Disruptor Screening Program. Environ. Health Perspect 2017, 125 (9), 096001. 10.1289/EHP1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Thomas RS; Bahadori T; Buckley TJ; Cowden J; Deisenroth C; Dionisio KL; Frithsen JB; Grulke CM; Gwinn MR; Harrill JA; Higuchi M; Houck KA; Hughes MF; Hunter ES; Isaacs KK; Judson RS; Knudsen TB; Lambert JC; Linnenbrink M; Martin TM; Newton SR; Padilla S; Patlewicz G; Paul-Friedman K; Phillips KA; Richard AM; Sams R; Shafer TJ; Setzer RW; Shah I; Simmons JE; Simmons SO; Singh A; Sobus JR; Strynar M; Swank A; Tornero-Valez R; Ulrich EM; Villeneuve DL; Wambaugh JF; Wetmore BA; Williams AJ The next Generation Blueprint of Computational Toxicology at the U.S. Environmental Protection Agency. Toxicol. Sci 2019. 10.1093/toxsci/kfz058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Harrill JA; Everett LJ; Haggard DE; Sheffield T; Bundy JL; Willis CM; Thomas RS; Shah I; Judson RS High-Throughput Transcriptomics Platform for Screening Environmental Chemicals. Toxicol. Sci 2021, 181 (1), 68–89. 10.1093/TOXSCI/KFAB009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Lee F; Shah I; Soong YT; Xing J; Ng IC; Tasnim F; Yu H Reproducibility and Robustness of High-Throughput S1500+ Transcriptomics on Primary Rat Hepatocytes for Chemical-Induced Hepatotoxicity Assessment. Curr. Res. Toxicol 2021. 10.1016/J.CRTOX.2021.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Yeakley JM; Shepard PJ; Goyena DE; VanSteenhouse HC; McComb JD; Seligmann BE A Trichostatin A Expression Signature Identified by TempO-Seq Targeted Whole Transcriptome Profiling. PLoS One 2017, 12 (5), e0178302. 10.1371/journal.pone.0178302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Musa A; Ghoraie LS; Zhang S-D; Galzko G; Yli-Harja O; Dehmer M; Haibe-Kains B; Emmert-Streib F A Review of Connectivity Map and Computational Approaches in Pharmacogenomics. Brief. Bioinform 2017, bbw112. 10.1093/bib/bbw112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Cheng J; Yang L; Kumar V; Agarwal P Systematic Evaluation of Connectivity Map for Disease Indications. Genome Med. 2014, 6 (12), 1–8. 10.1186/s13073-014-0095-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Wang Z; Gerstein M; Snyder M RNA-Seq: A Revolutionary Tool for Transcriptomics. Nat. Rev. Genet 2009, 10 (1), 57–63. 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Quackenbush J Microarray Data Normalization and Transformation. Nat. Genet 2002, 32 (Supp), 496–501. 10.1038/ng1032. [DOI] [PubMed] [Google Scholar]
- (75).Irizarry RA; Hobbs B; Collin F; Beazer-Barclay YD; Antonellis KJ; Scherf U; Speed TP Exploration, Normalization, and Summaries of High Density Oligonucleotide Array Probe Level Data. Biostatistics 2003, 4 (2), 249–264. 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- (76).Li C; Hung Wong W Model-Based Analysis of Oligonucleotide Arrays: Model Validation, Design Issues and Standard Error Application. Genome Biol. 2001, 2 (8), RESEARCH0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Subramanian A; Narayan R; Corsello SM; Peck DD; Natoli TE; Lu X; Gould J; Davis JF; Tubelli AA; Asiedu JK; Lahr DL; Hirschman JE; Liu Z; Donahue M; Julian B; Khan M; Wadden D; Smith IC; Lam D; Liberzon A; Toder C; Bagul M; Orzechowski M; Enache OM; Piccioni F; Johnson SA; Lyons NJ; Berger AH; Shamji AF; Brooks AN; Vrcic A; Flynn C; Rosains J; Takeda DY; Hu R; Davison D; Lamb J; Ardlie K; Hogstrom L; Greenside P; Gray NS; Clemons PA; Silver S; Wu X; Zhao W-N; Read-Button W; Wu X; Haggarty SJ; Ronco LV; Boehm JS; Schreiber SL; Doench JG; Bittker JA; Root DE; Wong B; Golub TR A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171 (6), 1437–1452.e17. 10.1016/J.CELL.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Conesa A; Madrigal P; Tarazona S; Gomez-Cabrero D; Cervera A; McPherson A; Szcześniak MW; Gaffney DJ; Elo LL; Zhang X; Mortazavi A A Survey of Best Practices for RNA-Seq Data Analysis. Genome Biol. 2016, 17 (1), 1–19. 10.1186/s13059-016-0881-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Thompson D; Botros I; Seligmann B Targeted Gene Expression Sequencing from FFPE: NPSeq™. In Cancer Research; American Association for Cancer Research (AACR), 2013; Vol. 73, pp 4146–4146. 10.1158/1538-7445.am2013-4146. [DOI] [Google Scholar]
- (80).Li H; Qiu J; Fu XD RASL-Seq for Massively Parallel and Quantitative Analysis of Gene Expression. Curr. Protoc. Mol. Biol 2012, CHAPTER (SUPPL.98), Unit4.13. 10.1002/0471142727.MB0413S98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Ye C; Ho DJ; Neri M; Yang C; Kulkarni T; Randhawa R; Henault M; Mostacci N; Farmer P; Renner S; Ihry R; Mansur L; Keller CG; McAllister G; Hild M; Jenkins J; Kaykas A DRUG-Seq for Miniaturized High-Throughput Transcriptome Profiling in Drug Discovery. Nat. Commun 2018, 9 (1), 1–9. 10.1038/s41467-018-06500-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Martin MT et al. Comparison of L1000 and Affymetrix Microarray for In Vitro Concentration-Response Gene Expression Profiling (SOT). Proceedings from the SOT annual meeting, March 22-26, 2015, San Diego, CA.EPA: Washington, D.C. 10.23645/EPACOMPTOX.5178763. [DOI] [Google Scholar]
- (83).Bushel PR; Paules RS; Auerbach SS A Comparison of the TempO-Seq S1500+ Platform to RNA-Seq and Microarray Using Rat Liver Mode of Action Samples. Front. Genet 2018, 9 (October), 1–14. 10.3389/fgene.2018.00485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Goytain A; Ng T NanoString NCounter Technology: High-Throughput RNA Validation. In Methods in Molecular Biology; Humana Press Inc., 2020; Vol. 2079, pp 125–139. 10.1007/978-1-4939-9904-0_10. [DOI] [PubMed] [Google Scholar]
- (85).Ritchie ME; Phipson B; Wu D; Hu Y; Law CW; Shi W; Smyth GK Limma Powers Differential Expression Analyses for RNA-Sequencing and Microarray Studies. Nucleic Acids Res. 2015, 43 (7), e47–e47. 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Anders S; Huber W Differential Expression Analysis for Sequence Count Data. Genome Biol. 2010, 11 (10), R106. 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Li J; Tibshirani R Finding Consistent Patterns: A Nonparametric Approach for Identifying Differential Expression in RNA-Seq Data. Stat. Methods Med. Res 2013, 22 (5), 519–536. 10.1177/0962280211428386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Johnson WE; Li C; Rabinovic A Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods. Biostatistics 2007, 8 (1), 118–127. 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- (89).Leek JT; Storey JD Capturing Heterogeneity in Gene Expression Studies by Surrogate Variable Analysis. PLoS Genet. 2007, 3 (9), 1724–1735. 10.1371/journal.pgen.0030161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Benito M; Parker J; Du Q; Wu J; Xiang D; Perou CM; Marron JS Adjustment of Systematic Microarray Data Biases. Bioinformatics 2004, 20 (1), 105–114. 10.1093/bioinformatics/btg385. [DOI] [PubMed] [Google Scholar]
- (91).Maglott D; Ostell J; Pruitt KD; Tatusova T Entrez Gene: Gene-Centered Information at NCBI. Nucleic Acids Res. 2011, 39 (SUPPL. 1), 52–57. 10.1093/nar/gkq1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Braschi B; Denny P; Gray K; Jones T; Seal R; Tweedie S; Yates B; Bruford E Genenames.Org: The HGNC and VGNC Resources in 2019. Nucleic Acids Res. 2019, 47 (D1), D786–D792. 10.1093/nar/gky930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Iorio F; Tagliaferri R; Bernardo D di. Identifying Network of Drug Mode of Action by Gene Expression Profiling. J. Comput. Biol 2009, 16 (2), 241–251. 10.1089/cmb.2008.10TT. [DOI] [PubMed] [Google Scholar]
- (94).Demir E; Cary MP; Paley S; Fukuda K; Lemer C; Vastrik I; Wu G; D’Eustachio P; Schaefer C; Luciano J; Schacherer F; Martinez-Flores I; Hu Z; Jimenez-Jacinto V; Joshi-Tope G; Kandasamy K; Lopez-Fuentes AC; Mi H; Pichler E; Rodchenkov I; Splendiani A; Tkachev S; Zucker J; Gopinath G; Rajasimha H; Ramakrishnan R; Shah I; Syed M; Anwar N; Babur Ö; Blinov M; Brauner E; Corwin D; Donaldson S; Gibbons F; Goldberg R; Hornbeck P; Luna A; Murray-Rust P; Neumann E; Reubenacker O; Samwald M; Van Iersel M; Wimalaratne S; Allen K; Braun B; Whirl-Carrillo M; Cheung K-H; Dahlquist K; Finney A; Gillespie M; Glass E; Gong L; Haw R; Honig M; Hubaut O; Kane D; Krupa S; Kutmon M; Leonard J; Marks D; Merberg D; Petri V; Pico A; Ravenscroft D; Ren L; Shah N; Sunshine M; Tang R; Whaley R; Letovksy S; Buetow KH; Rzhetsky A; Schachter V; Sobral BS; Dogrusoz U; McWeeney S; Aladjem M; Birney E; Collado-Vides J; Goto S; Hucka M; Novère NL; Maltsev N; Pandey A; Thomas P; Wingender E; Karp PD; Sander C; Bader GD The BioPAX Community Standard for Pathway Data Sharing. Nat. Biotechnol 2010, 28 (9). 10.1038/nbt.1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Koleti A; Terryn R; Stathias V; Chung C; Cooper DJ; Turner JP; Vidović D; Forlin M; Kelley TT; D’Urso A; Allen BK; Torre D; Jagodnik KM; Wang L; Jenkins SL; Mader C; Niu W; Fazel M; Mahi N; Pilarczyk M; Clark N; Shamsaei B; Meller J; Vasiliauskas J; Reichard J; Medvedovic M; Ma’ayan A; Pillai A; Schürer SC Data Portal for the Library of Integrated Network-Based Cellular Signatures (LINCS) Program: Integrated Access to Diverse Large-Scale Cellular Perturbation Response Data. Nucleic Acids Res. 2018, 46 (D1), D558–D566. 10.1093/nar/gkx1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (96).Affymetrix. Affymetrix Support by Product for GeneChip® Human Genome U133 Plus 2.0 Array. http://www.affymetrix.com/support/technical/byproduct.affx?product=hg-u133-plus (accessed 2018-07-26).
- (97).Brazma A; Hingamp P; Quackenbush J; Sherlock G; Spellman P; Stoeckert C; Aach J; Ansorge W; Ball CA; Causton HC; Gaasterland T; Glenisson P; Holstege FCP; Kim IF; Markowitz V; Matese JC; Parkinson H; Robinson A; Sarkans U; Schulze-Kremer S; Stewart J; Taylor R; Vilo J; Vingron M Minimum Information about a Microarray Experiment (MIAME)-toward Standards for Microarray Data. Nat. Genet 2001, 29 (4), 365–371. 10.1038/ng1201-365. [DOI] [PubMed] [Google Scholar]
- (98).Team RCR: A Language and Environment for Statistical Computing. Vienna, Austria: 2014. [Google Scholar]
- (99).Davis S; Meltzer PS GEOquery: A Bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics 2007, 23 (14), 1846–1847. 10.1093/bioinformatics/btm254. [DOI] [PubMed] [Google Scholar]
- (100).Cock PJA; Antao T; Chang JT; Chapman BA; Cox CJ; Dalke A; Friedberg I; Hamelryck T; Kauff F; Wilczynski B; de Hoon MJL Biopython: Freely Available Python Tools for Computational Molecular Biology and Bioinformatics. Bioinformatics 2009, 25 (11), 1422–1423. 10.1093/bioinformatics/btp163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Brazma A; Parkinson H; Sarkans U; Shojatalab M; Vilo J; Abeygunawardena N; Holloway E; Kapushesky M; Kemmeren P; Lara GG; Oezcimen A; Rocca-Serra P; Sansone SA ArrayExpress—a Public Repository for Microarray Gene Expression Data at the EBI. Nucleic Acids Res. 2003, 31 (1), 68–71. 10.1093/NAR/GKG091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Kolesnikov N; Hastings E; Keays M; Melnichuk O; Tang YA; Williams E; Dylag M; Kurbatova N; Brandizi M; Burdett T; Megy K; Pilicheva E; Rustici G; Tikhonov A; Parkinson H; Petryszak R; Sarkans U; Brazma A ArrayExpress Update—Simplifying Data Submissions. Nucleic Acids Res. 2015, 43 (D1), D1113–D1116. 10.1093/nar/gku1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Malone J; Holloway E; Adamusiak T; Kapushesky M; Zheng J; Kolesnikov N; Zhukova A; Brazma A; Parkinson H Modeling Sample Variables with an Experimental Factor Ontology. Bioinformatics 2010, 26 (8), 1112–1118. 10.1093/BIOINFORMATICS/BTQ099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Okido T; Kodama Y; Mashima J; Kosuge T; Fujisawa T; Ogasawara O DNA Data Bank of Japan (DDBJ) Update Report 2021. Nucleic Acids Res. 2022, 50 (D1), D102–D105. 10.1093/NAR/GKAB995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Bono H All of Gene Expression (AOE): An Integrated Index for Public Gene Expression Databases. PLoS One 2020, 15 (1), e0227076. 10.1371/JOURNAL.PONE.0227076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Kauffmann A; Rayner TF; Parkinson H; Kapushesky M; Lukk M; Brazma A; Huber W Importing ArrayExpress Datasets into R/Bioconductor. Bioinformatics 2009, 25 (16), 2092–2094. 10.1093/BIOINFORMATICS/BTP354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Cokelaer T; Pultz D; Harder LM; Serra-Musach J; Saez-Rodriguez J; Valencia A BioServices: A Common Python Package to Access Biological Web Services Programmatically. Bioinformatics 2013, 29 (24), 3241–3242. 10.1093/bioinformatics/btt547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Engreitz JM; Chen R; Morgan AA; Dudley JT; Mallelwar R; Butte AJ ProfileChaser: Searching Microarray Repositories Based on Genome-Wide Patterns of Differential Expression. Bioinformatics 2011, 27 (23), 3317–3318. 10.1093/bioinformatics/btr548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Fujibuchi W; Kiseleva L; Taniguchi T; Harada H; Horton P CellMontage: Similar Expression Profile Search Server. Bioinformatics 2007, 23 (22), 3103–3104. 10.1093/bioinformatics/btm462. [DOI] [PubMed] [Google Scholar]
- (110).Chang JT; Gatza ML; Lucas JE; Barry WT; Vaughn P; Nevins JR SIGNATURE: A Workbench for Gene Expression Signature Analysis. BMC Bioinformatics 2011, 12 (1), 443. 10.1186/1471-2105-12-443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Williams G A Searchable Cross-Platform Gene Expression Database Reveals Connections between Drug Treatments and Disease. BMC Genomics 2012, 13 (1), 12. 10.1186/1471-2164-13-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Wang Z; Monteiro CD; Jagodnik KM; Fernandez NF; Gundersen GW; Rouillard AD; Jenkins SL; Feldmann AS; Hu KS; McDermott MG; Duan Q; Clark NR; Jones MR; Kou Y; Goff T; Woodland H; Amaral FMR; Szeto GL; Fuchs O; Schüssler-Fiorenza Rose SM; Sharma S; Schwartz U; Bausela XB; Szymkiewicz M; Maroulis V; Salykin A; Barra CM; Kruth CD; Bongio NJ; Mathur V; Todoric RD; Rubin UE; Malatras A; Fulp CT; Galindo JA; Motiejunaite R; Jüschke C; Dishuck PC; Lahl K; Jafari M; Aibar S; Zaravinos A; Steenhuizen LH; Allison LR; Gamallo P; de Andres Segura F; Dae Devlin T; Pérez-García V; Ma’ayan A Extraction and Analysis of Signatures from the Gene Expression Omnibus by the Crowd. Nat. Commun 2016, 7, 12846. 10.1038/ncomms12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Liberzon A; Subramanian A; Pinchback R; Thorvaldsdóttir H; Tamayo P; Mesirov JP Molecular Signatures Database (MSigDB) 3.0. Bioinformatics 2011, 27 (12), 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Liberzon A; Birger C; Thorvaldsdóttir H; Ghandi M; Mesirov JP; Tamayo P The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 2015, 1 (6), 417–425. 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Jassal B; Matthews L; Viteri G; Gong C; Lorente P; Fabregat A; Sidiropoulos K; Cook J; Gillespie M; Haw R; Loney F; May B; Milacic M; Rothfels K; Sevilla C; Shamovsky V; Shorser S; Varusai T; Weiser J; Wu G; Stein L; Hermjakob H; D’Eustachio P The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2020, 48 (D1), D498–D503. 10.1093/nar/gkz1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Kanehisa M Toward Understanding the Origin and Evolution of Cellular Organisms. Protein Sci. 2019, 28 (11), 1947–1951. 10.1002/pro.3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Schaefer CF; Anthony K; Krupa S; Buchoff J; Day M; Hannay T; Buetow KH PID: The Pathway Interaction Database. Nucleic Acids Res. 2009, 37 (SUPPL. 1), 674–679. 10.1093/nar/gkn653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Carbon S; Douglass E; Dunn N; Good B; Harris NL; Lewis SE; Mungall CJ; Basu S; Chisholm RL; Dodson RJ; Hartline E; Fey P; Thomas PD; Albou LP; Ebert D; Kesling MJ; Mi H; Muruganujan A; Huang X; Poudel S; Mushayahama T; Hu JC; LaBonte SA; Siegele DA; Antonazzo G; Attrill H; Brown NH; Fexova S; Garapati P; Jones TEM; Marygold SJ; Millburn GH; Rey AJ; Trovisco V; Dos Santos G; Emmert DB; Falls K; Zhou P; Goodman JL; Strelets VB; Thurmond J; Courtot M; Osumi DS; Parkinson H; Roncaglia P; Acencio ML; Kuiper M; Lreid A; Logie C; Lovering RC; Huntley RP; Denny P; Campbell NH; Kramarz B; Acquaah V; Ahmad SH; Chen H; Rawson JH; Chibucos MC; Giglio M; Nadendla S; Tauber R; Duesbury MJ; Del NT; Meldal BHM; Perfetto L; Porras P; Orchard S; Shrivastava A; Xie Z; Chang HY; Finn RD; Mitchell AL; Rawlings ND; Richardson L; Sangrador-Vegas A; Blake JA; Christie KR; Dolan ME; Drabkin HJ; Hill DP; Ni L; Sitnikov D; Harris MA; Oliver SG; Rutherford K; Wood V; Hayles J; Bahler J; Lock A; Bolton ER; De Pons J; Dwinell M; Hayman GT; Laulederkind SJF; Shimoyama M; Tutaj M; Wang SJ; D’Eustachio P; Matthews L; Balhoff JP; Aleksander SA; Binkley G; Dunn BL; Cherry JM; Engel SR; Gondwe F; Karra K; MacPherson KA; Miyasato SR; Nash RS; Ng PC; Sheppard TK; Shrivatsav Vp A; Simison M; Skrzypek MS; Weng S; Wong ED; Feuermann M; Gaudet P; Bakker E; Berardini TZ; Reiser L; Subramaniam S; Huala E; Arighi C; Auchincloss A; Axelsen K; Argoud GP; Bateman A; Bely B; Blatter MC; Boutet E; Breuza L; Bridge A; Britto R; Bye-A-Jee H; Casals-Casas C; Coudert E; Estreicher A; Famiglietti L; Garmiri P; Georghiou G; Gos A; Gruaz-Gumowski N; Hatton-Ellis E; Hinz U; Hulo C; Ignatchenko A; Jungo F; Keller G; Laiho K; Lemercier P; Lieberherr D; Lussi Y; Mac-Dougall A; Magrane M; Martin MJ; Masson P; Natale DA; Hyka NN; Pedruzzi I; Pichler K; Poux S; Rivoire C; Rodriguez-Lopez M; Sawford T; Speretta E; Shypitsyna A; Stutz A; Sundaram S; Tognolli M; Tyagi N; Warner K; Zaru R; Wu C; Chan J; Cho J; Gao S; Grove C; Harrison MC; Howe K; Lee R; Mendel J; Muller HM; Raciti D; Van Auken K; Berriman M; Stein L; Sternberg PW; Howe D; Toro S; Westerfield M The Gene Ontology Resource: 20 Years and Still GOing Strong. Nucleic Acids Res. 2019, 47 (D1), D330–D338. 10.1093/nar/gky1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Irizarry RA; Wang C; Zhou Y; Speed TP Gene Set Enrichment Analysis Made Simple. Stat. Methods Med. Res 2009, 18 (6), 565–575. 10.1177/0962280209351908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Tanner SW; Agarwal P Gene Vector Analysis (Geneva): A Unified Method to Detect Differentially-Regulated Gene Sets and Similar Microarray Experiments. BMC Bioinformatics 2008, 9, 348. 10.1186/1471-2105-9-348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Chung FH; Chiang YR; Tseng AL; Sung YC; Lu J; Huang MC; Ma N; Lee HC Functional Module Connectivity Map (FMCM): A Framework for Searching Repurposed Drug Compounds for Systems Treatment of Cancer and an Application to Colorectal Adenocarcinoma. PLoS One 2014, 9 (1). 10.1371/journal.pone.0086299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Yi Y; Li C; Miller C; George AL Jr. Strategy for Encoding and Comparison of Gene Expression Signatures. Genome Biol. 2007, 8 (7), R133. 10.1186/gb-2007-8-7-r133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Tian L; Greenberg SA; Kong SW; Altschuler J; Kohane IS; Park PJ Discovering Statistically Significant Pathways in Expression Profiling Studies. Proc. Natl. Acad. Sci. U. S. A 2005, 102 (38), 13544–13549. 10.1073/pnas.0506577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Goeman JJ; van de Geer SA; de Kort F; van Houwelingen HC A Global Test for Groups of Genes: Testing Association with a Clinical Outcome. Bioinformatics 2004, 20 (1), 93–99. 10.1093/bioinformatics/btg382. [DOI] [PubMed] [Google Scholar]
- (125).Goeman JJ; Oosting J; Cleton-Jansen A-M; Anninga JK; van Houwelingen HC Testing Association of a Pathway with Survival Using Gene Expression Data. Bioinformatics 2005, 21 (9), 1950–1957. 10.1093/bioinformatics/bti267. [DOI] [PubMed] [Google Scholar]
- (126).Al-Shahrour F; Diaz-Uriarte R; Dopazo J Discovering Molecular Functions Significantly Related to Phenotypes by Combining Gene Expression Data and Biological Information. Bioinformatics 2005, 21 (13), 2988–2993. 10.1093/bioinformatics/bti457. [DOI] [PubMed] [Google Scholar]
- (127).Kim S-Y; Volsky DJ PAGE: Parametric Analysis of Gene Set Enrichment. BMC Bioinformatics 2005, 6 (1), 144. 10.1186/1471-2105-6-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Sartor MA; Leikauf GD; Medvedovic M LRpath: A Logistic Regression Approach for Identifying Enriched Biological Groups in Gene Expression Data. Bioinformatics 2009, 25 (2), 211–217. 10.1093/bioinformatics/btn592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).Newton MA; Quintana FA; den Boon JA; Sengupta S; Ahlquist P Random-Set Methods Identify Distinct Aspects of the Enrichment Signal in Gene-Set Analysis. Ann. Appl. Stat 2007, 1 (1), 85–106. 10.1214/07-AOAS104. [DOI] [Google Scholar]
- (130).Barry WT; Nobel AB; Wright FA Significance Analysis of Functional Categories in Gene Expression Studies: A Structured Permutation Approach. Bioinformatics 2005, 21 (9), 1943–1949. 10.1093/bioinformatics/bti260. [DOI] [PubMed] [Google Scholar]
- (131).Gower AC; Spira A; Lenburg ME Discovering Biological Connections between Experimental Conditions Based on Common Patterns of Differential Gene Expression. BMC Bioinformatics 2011, 12 (1), 381. 10.1186/1471-2105-12-381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Efron B; Tibshirani R On Testing the Significance of Sets of Genes. Ann. Appl. Stat 2007, 1 (1), 107–129. 10.1214/07-AOAS101. [DOI] [Google Scholar]
- (133).Zhang S-D; Gant TW SscMap: An Extensible Java Application for Connecting Small-Molecule Drugs Using Gene-Expression Signatures. BMC Bioinformatics 2009, 10 (1), 236. 10.1186/1471-2105-10-236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Hänzelmann S; Castelo R; Guinney J GSVA: Gene Set Variation Analysis for Microarray and RNA-Seq Data. BMC Bioinformatics 2013, 14. 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Iskar M; Campillos M; Kuhn M; Jensen LJ; van Noort V; Bork P Drug-Induced Regulation of Target Expression. PLoS Comput. Biol 2010, 6 (9). 10.1371/journal.pcbi.1000925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (136).Cha S Comprehensive Survey on Distance / Similarity Measures between Probability Density Functions. Int. J. Math. Model. Methods Appl. Sci 2007, 1 (4), 300–307. 10.1007/s00167-009-0884-z. [DOI] [Google Scholar]
- (137).Cheng J; XIE Q; KUMAR V; HURLE M; FREUDENBERG JM; YANG L; AGARWAL P Evaluation of Analytical Methods for Connectivity Map Data. Biocomput. 2013. 2012, 5–16. 10.1142/9789814447973_0002. [DOI] [PubMed] [Google Scholar]
- (138).Zhang SD A Simple and Robust Method for Connecting Small-Molecule Drugs Using Gene-Expression Signatures. BMC Bioinformatics 2008, 9, 1–10. 10.1186/1471-2105-9-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (139).Jaccard P Lois de Distribution Florale Dans La Zone Alpine. Bull. la Société Vaudoise des Sci. Nat 1902, 38, 67–130. 10.5169/seals-266762. [DOI] [Google Scholar]
- (140).Goeman JJ; Buhlmann P Analyzing Gene Expression Data in Terms of Gene Sets: Methodological Issues. Bioinformatics 2007, 23 (8), 980–987. 10.1093/bioinformatics/btm051. [DOI] [PubMed] [Google Scholar]
- (141).Keenan AB; Jenkins SL; Jagodnik KM; Koplev S; He E; Torre D; Wang Z; Dohlman AB; Silverstein MC; Lachmann A; Kuleshov MV; Ma’ayan A; Stathias V; Terryn R; Cooper D; Forlin M; Koleti A; Vidovic D; Chung C; Schürer SC; Vasiliauskas J; Pilarczyk M; Shamsaei B; Fazel M; Ren Y; Niu W; Clark NA; White S; Mahi N; Zhang L; Kouril M; Reichard JF; Sivaganesan S; Medvedovic M; Meller J; Koch RJ; Birtwistle MR; Iyengar R; Sobie EA; Azeloglu EU; Kaye J; Osterloh J; Haston K; Kalra J; Finkbiener S; Li J; Milani P; Adam M; Escalante-Chong R; Sachs K; Lenail A; Ramamoorthy D; Fraenkel E; Daigle G; Hussain U; Coye A; Rothstein J; Sareen D; Ornelas L; Banuelos M; Mandefro B; Ho R; Svendsen CN; Lim RG; Stocksdale J; Casale MS; Thompson TG; Wu J; Thompson LM; Dardov V; Venkatraman V; Matlock A; Van Eyk JE; Jaffe JD; Papanastasiou M; Subramanian A; Golub TR; Erickson SD; Fallahi-Sichani M; Hafner M; Gray NS; Lin JR; Mills CE; Muhlich JL; Niepel M; Shamu CE; Williams EH; Wrobel D; Sorger PK; Heiser LM; Gray JW; Korkola JE; Mills GB; LaBarge M; Feiler HS; Dane MA; Bucher E; Nederlof M; Sudar D; Gross S; Kilburn DF; Smith R; Devlin K; Margolis R; Derr L; Lee A; Pillai A The Library of Integrated Network-Based Cellular Signatures NIH Program: System-Level Cataloging of Human Cells Response to Perturbations. Cell Syst. 2018, 6 (1), 13–24. 10.1016/j.cels.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).World Health Organization. The Anatomical Therapeutic Chemical Classification System with Defined Daily Doses (ATC/DDD). http://www.who.int/classifications/atcddd/en/ (accessed 2018-08-13). [Google Scholar]
- (143).Judson R; Houck K; Martin M; Knudsen T; Thomas RS; Sipes N; Shah I; Wambaugh J; Crofton K In Vitro and Modelling Approaches to Risk Assessment from the U.S. Environmental Protection Agency ToxCast Programme. Basic Clin. Pharmacol. Toxicol 2014, 115 (1), 69–76. 10.1111/bcpt.12239. [DOI] [PubMed] [Google Scholar]
- (144).Richard AM; Judson RS; Houck KA; Grulke CM; Volarath P; Thillainadarajah I; Yang C; Rathman J; Martin MT; Wambaugh JF; Knudsen TB; Kancherla J; Mansouri K; Patlewicz G; Williams AJ; Little SB; Crofton KM; Thomas RS ToxCast Chemical Landscape: Paving the Road to 21st Century Toxicology. Chemical Research in Toxicology. 2016. 10.1021/acs.chemrestox.6b00135. [DOI] [PubMed] [Google Scholar]
- (145).USEPA. Directive to Prioritize Efforts to Reduce Animal Testing. US Environmental Protection Agency; 2019. [Google Scholar]
- (146).USEPA. EPA New Approach Methods Work Plan: Reducing Use of Animals in Chemical Testing. 2020.
- (147).Wang R-L; Biales AD; Garcia-Reyero N; Perkins EJ; Villeneuve DL; Ankley GT; Bencic DC Fish Connectivity Mapping: Linking Chemical Stressors by Their Mechanisms of Action-Driven Transcriptomic Profiles. BMC Genomics 2016, 17, 84. 10.1186/s12864-016-2406-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Chambers B; Shah I Elucidating Stress Response Pathway Activity Using Transcriptomics. Comput. Toxicol 2021, (submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Williams AJ; Grulke CM; Edwards J; McEachran AD; Mansouri K; Baker NC; Patlewicz G; Shah I; Wambaugh JF; Judson RS; Richard AM The CompTox Chemistry Dashboard: A Community Data Resource for Environmental Chemistry. J. Cheminform 2017, 9 (1). 10.1186/s13321-017-0247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Judson RS; Thomas RS; Baker N; Simha A; Howey XM; Marable C; Kleinstreuer NC; Houck KA Workflow for Defining Reference Chemicals for Assessing Performance of in Vitro Assays. ALTEX 2019, 36 (2), 261–276. 10.14573/altex.1809281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Chambers B; Basili D; Word L; Baker N; Middleton A; Judson R; Shah I Searching for LINCS to Stress: Literature-Linked Transcriptomic Analysis Identifies Stress Response Active Chemical Targets. (in-preparation) 2022. [Google Scholar]
- (152).Shi L; Reid LH; Jones WD; Shippy R; Warrington JA; Baker SC; Collins PJ; Longueville F, de; Kawasaki ES; Lee KY; Luo Y; Sun YA; Willey JC; Setterquist RA; Fischer GM; Tong W; Dragan YP; Dix DJ; Frueh FW; Goodsaid FM; Herman D; Jensen RV; Johnson CD; Lobenhofer EK; Puri RK; Scherf U; Thierry-Mieg J; Wang C; Wilson M; Wolber PK; Zhang L; Amur S; Bao W; Barbacioru CC; Lucas AB; Bertholet V; Boysen C; Bromley B; Brown D; Brunner A; Canales R; Cao XM; Cebula TA; Chen JJ; Cheng J; Chu T-M; Chudin E; Corson J; Corton JC; Croner LJ; Davies C; Davison TS; Delenstarr G; Deng X; Dorris D; Eklund AC; Fan X; Fang H; Fulmer-Smentek S; Fuscoe JC; Gallagher K; W. Ge; Guo L; Guo X; Hager J; Haje PK; Han J; Han T; Harbottle HC; Harris SC; Hatchwell E; Hauser CA; Hester S; Hong H; Hurban P; Jackson SA; Ji H; Knight CR; Kuo WP; LeClerc JE; Levy S; Li Q-Z; Liu C; Liu Y; Lombardi MJ; Ma Y; Magnuson SR; Maqsodi B; McDaniel T; Mei N; Myklebost O; Ning B; Novoradovskaya N; Orr MS; Osborn TW; Papallo A; Patterson TA; Perkins RG; Peters EH; Peterson R; Philips KL; Pine PS; Pusztai L; Qian F; Ren H; Rosen M; Rosenzweig BA; Samaha RR; Schena M; Schroth GP; Shchegrova S; Smith DD; Staedtler F; Su Z; Sun H; Szallasi Z; Tezak Z; Thierry-Mieg D; Thompson KL; Tikhonova I; Turpaz Y; Vallanat B; Van C; Walker SJ; Wang SJ; Wang Y; Wolfinger R; Wong A; Wu J; Xiao C; Xie Q; Xu J; Yang W; Zhang L; Zhong S; Zong Y; S. W Jr, The MicroArray Quality Control (MAQC) Project Shows Inter- and Intraplatform Reproducibility of Gene Expression Measurements. Nat. Biotechnol 2006, 24 (9), 1151–1161. 10.1038/nbt1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Villeneuve DL; Crump D; Garcia-Reyero N; Hecker M; Hutchinson TH; LaLone CA; Landesmann B; Lettieri T; Munn S; Nepelska M; Ottinger MA; Vergauwen L; Whelan M Adverse Outcome Pathway (AOP) Development I: Strategies and Principles. Toxicol. Sci 2014, 142 (2), 312–320. 10.1093/toxsci/kfu199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).US EPA. The Toxic Substances Control Act (TSCA) Chemical Substance Inventory. https://www.epa.gov/tsca-inventory (accessed 2019-08-05).
- (155).Chambers B; Shah I Evaluating Adaptive Stress Response Gene Signatures Using Transcriptomics. Comput. Toxicol 2021, 20, 100179. 10.1016/J.COMTOX.2021.100179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (156).Nuwaysir EF; Bittner M; Trent J; Barrett JC; Afshari CA Microarrays and Toxicology: The Advent of Toxicogenomics. Mol. Carcinog 1999, 24 (3), 153–159. . [DOI] [PubMed] [Google Scholar]
- (157).Fielden MR; Zacharewski TR Challenges and Limitations of Gene Expression Profiling in Mechanistic and Predictive Toxicology. Toxicol. Sci 2001, 60 (1), 6–10. 10.1093/toxsci/60.1.6. [DOI] [PubMed] [Google Scholar]
- (158).Hansen KD; Wu Z; Irizarry RA; Leek JT Sequencing Technology Does Not Eliminate Biological Variability. Nat. Biotechnol 2011, 29 (7), 572–573. 10.1038/nbt.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Wenric S; Shemirani R Using Supervised Learning Methods for Gene Selection in RNA-Seq Case-Control Studies. Front. Genet 2018, 9, 297. 10.3389/fgene.2018.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Igarashi Y; Nakatsu N; Yamashita T; Ono A; Ohno Y; Urushidani T; Yamada H Open TG-GATEs: A Large-Scale Toxicogenomics Database. Nucleic Acids Res. 2015, 43 (D1), D921–D927. 10.1093/nar/gku955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Parkkinen JA; Kaski S Probabilistic Drug Connectivity Mapping. BMC Bioinformatics 2014, 15 (1), 1–10. 10.1186/1471-2105-15-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (162).Virtanen S; Klami A; Khan SA; Kaski S Bayesian Group Factor Analysis. 2011, XX. 10.1016/j.neunet.2007.12.026. [DOI] [PubMed] [Google Scholar]
- (163).Carvalho CM; Chang J; Lucas JE; Nevins JR; Wang Q; West M High-Dimensional Sparse Factor Modeling: Applications in Gene Expression Genomics. J. Am. Stat. Assoc 2008, 103 (484), 1438–1456. 10.1198/016214508000000869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Lecun Y; Bengio Y; Hinton G Deep Learning. Nature 2015, 521 (7553), 436–444. 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- (165).Chen HIH; Chiu YC; Zhang T; Zhang S; Huang Y; Chen Y GSAE: An Autoencoder with Embedded Gene-Set Nodes for Genomics Functional Characterization. BMC Syst. Biol 2018, 12 (8), 45–57. 10.1186/S12918-018-0642-2/TABLES/8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Magnusson R; Tegnér JN; Gustafsson M Deep Neural Network Prediction of Genome-Wide Transcriptome Signatures – beyond the Black-Box. npj Syst. Biol Appl. 2022 81 2022, 8 (1), 1–8. 10.1038/s41540-022-00218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (167).Iorio F; Saez-Rodriguez J; di Bernardo D Network Based Elucidation of Drug Response: From Modulators to Targets. BMC Syst. Biol 2013, 7, 139. 10.1186/1752-0509-7-139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Liu A; Trairatphisan P; Gjerga E; Didangelos A; Barratt J; Saez-Rodriguez J From Expression Footprints to Causal Pathways: Contextualizing Large Signaling Networks with CARNIVAL. npj Syst. Biol. Appl 2019, 5 (1), 1–10. 10.1038/s41540-019-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Denes T; Valdeolivas A; Gul L; as Palacio-Escat N; Klein M; Ivanova O; arton Ölbei M; Abor AG; Theis F; Odos DM; as Korcsm aros T; Saez-Rodriguez J Integrated Intra- and Intercellular Signaling Knowledge for Multicellular Omics Analysis. Mol. Syst. Biol 2021, 17 (3), e9923. 10.15252/MSB.20209923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Zickenrott S; Angarica VE; Upadhyaya BB; del Sol A Prediction of Disease–Gene–Drug Relationships Following a Differential Network Analysis. Cell Death Dis. 2016, 7 (1), e2040. 10.1038/cddis.2015.393. [DOI] [PMC free article] [PubMed] [Google Scholar]