
Abstract 抽象的
High-throughput single-cell profiling provides an unprecedented ability to uncover the molecular states of millions of cells. These technologies are, however, destructive to cells and tissues, raising practical challenges when aiming to track dynamic biological processes. As the same cell cannot be observed at multiple time points, as it changes in time and space in response to a stimulus or perturbation, these large-scale measurements only produce unaligned data sets. In this Primer, we show how such challenges can be effectively addressed using the unifying framework of optimal transport theory and tackled using the many algorithms that have been proposed for the range of scenarios of key interest in computational biology. We further review recent advances integrating optimal transport and deep learning that allow forecasting heterogeneous cellular dynamics and behaviour, crucial in particular for pressing problems in personalized medicine.
高通量单细胞分析提供了前所未有的能力,可以揭示数百万个细胞的分子状态。然而,这些技术对细胞和组织具有破坏性,在追踪动态生物过程时带来了实际挑战。由于同一个细胞会随着刺激或扰动而随时间和空间发生变化,因此无法在多个时间点进行观察,这些大规模测量只能产生不一致的数据集。在本入门指南中,我们将展示如何使用最优传输理论的统一框架有效地应对此类挑战,并使用针对计算生物学中一系列关键场景提出的众多算法来应对。我们还将进一步回顾整合最优传输和深度学习的最新进展,这些进展可以预测异质细胞动力学和行为,这对于个性化医疗中亟待解决的问题尤其重要。
Similar content being viewed by others
其他人正在查看类似内容
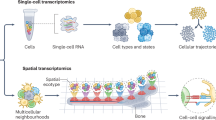

Introduction 介绍
Biological systems are dynamic at multiple scales, ranging from molecular and cellular to tissue, organ and organismal behaviour. At the cellular level, single-cell omics now provide a direct window into the molecular makeup of individual cells, which is both comprehensive and has high resolution, allowing us to capture a detailed snapshot of the molecular state of cells at a given point in time. Similarly, at the tissue level, advances in imaging and spatial omics help map cells and their molecular state as they are geometrically organized in tissues, improving our understanding of key physiological processes.
生物系统在多个尺度上都是动态的,涵盖从分子和细胞到组织、器官和生物体行为的各个层面。在细胞层面,单细胞组学如今提供了一个直接了解单个细胞分子组成的窗口,它不仅全面,而且分辨率高,使我们能够捕捉到特定时间点细胞分子状态的详细快照。同样,在组织层面,成像和空间组学的进步有助于绘制细胞及其在组织中几何排列的分子状态图,从而加深我们对关键生理过程的理解。
Although single-cell and spatial methods can now routinely generate millions of cell profiles, they do come with an important limitation: these methods are destructive assays, such that the same cell cannot be observed twice, and hence the resulting data are not aligned. This limitation has acute implications both for studying basic biological systems, from developmental biology to immunology, and in translational research, when we aim to monitor the response during pathogenesis or under treatment across multiple patients. Because of the destructive nature, the same cell cannot be profiled multiple times along a time course or measured using multiple modalities (unless they are measured simultaneously). In the case of single-cell technologies, physical dissociation of tissues also leads to loss of spatial information. To relate time points, modalities or positions require us to align and connect profiles collected from different cells post hoc.
尽管单细胞和空间方法现在可以常规生成数百万个细胞图谱,但它们也有一个重要的局限性:这些方法是破坏性测定,即无法对同一个细胞进行两次观察,因此得到的数据不一致。这种局限性对于研究从发育生物学到免疫学的基础生物系统以及在转化研究中(当我们旨在监测多个患者在发病过程中或治疗过程中的反应时)都具有严重的影响。由于破坏性,同一个细胞不能沿着时间进程进行多次分析,也不能使用多种模态进行测量(除非同时测量)。对于单细胞技术,组织的物理解离也会导致空间信息的丢失。为了关联时间点、模态或位置,我们需要事后对齐和连接从不同细胞收集的图谱。
The need to realign data sets is a common thread to all of these problems. Accordingly, a unifying mathematical framework, optimal transport (OT)1,2, has emerged as an important solution. OT theory is a major research area in pure mathematics (see works by Villani1, Figalli3 and Caffarelli4 for examples), which has been adopted as an approach to fill this gap in single-cell and spatial omics in silico. OT reconstructs how a source population (represented as one probability distribution) can morph efficiently into another target population, given only source and target samples. For example, if the source distribution is a sample of pre-stimulation cells, and the target is another sample of cells at some time point post-stimulation, OT can reconstruct the unobserved temporal process and provide an informed guess to recover an OT map relating the two cell populations and reconstructing the effect of the stimulation5,6. With the development of deep learning parameterizations of OT, neural OT now allows us to predict how a perturbation might affect previously unseen cells — such as a different cell type stimulated in the same way or the cells of another patient with the same disease6,7,8 — opening important opportunities in the field of precision medicine (Fig. 1).
重新调整数据集的需求是所有这些问题的共同点。因此,一个统一的数学框架, 最佳传输 (OT) 1、2 , 已成为重要的解决方案。OT 理论是纯数学中的一个主要研究领域(例如,参见 Villani 1 、Figalli 3 和 Caffarelli 4 的作品),它已被用作填补单细胞和空间组学计算机模拟中这一空白的方法。OT 重建了在仅给定源样本和目标样本的情况下,源群体(表示为一个概率分布)如何有效地转变为另一个目标群体。例如,如果源分布是刺激前细胞的样本,而目标是刺激后某个时间点的另一个细胞样本,则 OT 可以重建未观察到的时间过程并提供有根据的猜测来恢复与两个细胞群相关的 OT 图并重建刺激的效果 5、6 。 随着 OT 深度学习参数化的发展,神经 OT 现在使我们能够预测扰动如何影响以前未见过的细胞(例如以相同方式刺激的不同细胞类型或患有相同疾病的另一个患者的细胞 6、7、8 ) , 这为精准医疗领域开辟了重要机遇(图 1 )。
图 1:单细胞和空间生物学中最佳运输的概述。

Optimal transport (OT) finds various use cases in single-cell biology. a, In the reconstruction of cellular differentiation processes in developmental biology, OT provides alignment among consecutive measurements and thus infers progenitors and descendants of each cell x. b, The re-alignment of single-cell measurements of each cell population before μ and after a perturbation ν, such as a treatment, chemical or genetic intervention. OT enables the reconstruction of fine-grained perturbation responses of heterogeneous cell populations222,223,224. Parameterizing the OT with neural networks also allows the use of OT in predicting treatment outcomes of unseen cells, such as those from a new cell type or patient. c, OT can be employed to spatially reconstruct single-cell data. Given a reference atlas or spatially resolved single-cell measurements, OT is able to restore tissue geometries and architectures of single cells recorded using non-spatially-resolved measurement technologies. d, Advances in the development of high-throughput measurement technologies facilitate the recording of a biological system using different data modalities. OT can facilitate re-aligning measurements across modalities to provide a diverse characterization of similar cell states. RNA-seq, RNA sequencing.
最佳传输 (OT) 在单细胞生物学中有多种用途。a 、 在发育生物学中重建细胞分化过程时,OT 提供连续测量之间的比对,从而推断每个细胞 x 的祖细胞和后代 。b 、在 μ 之前和扰动 ν 之后(例如治疗、化学或基因干预)重新调整每个细胞群的单细胞测量值。OT 能够重建异质细胞群 222、223、224 的细粒度扰动响应。使用神经网络参数化 OT 还允许使用 OT 预测看不见的细胞(例如来自新细胞类型或患者的细胞)的治疗结果 。c 、OT 可用于空间重建单细胞数据。给定参考图谱或空间分辨的单细胞测量值,OT 能够恢复使用非空间分辨测量技术记录的单细胞的组织几何形状和结构。 d 、高通量测量技术的进步促进了使用不同数据模式记录生物系统。OT 可以促进跨模式重新调整测量,从而提供相似细胞状态的多样化表征。RNA-seq,RNA 测序。
In single-cell biology, OT has been used to infer the distributions of ancestors and descendants of cells along developmental processes5 (Fig. 1a), perform trajectory inference5,9,10,11,12,13,14,15,16, predict perturbation responses6,17,18 (Fig. 1b), spatially reconstruct the positions of cells in tissues19,20 (Fig. 1c), integrate multi-omics data of different molecular modalities21 (Fig. 1d), infer cell–cell similarity22, integrate across scales or views (for example, in morphology and molecular profiling)23 as well as missing modality imputation24.
在单细胞生物学中,OT 已用于推断细胞祖先和后代在发育过程中的分布 5 (图 1a ) , 执行轨迹推断 5、9、10、11、12、13、14、15、16 , 预测扰动响应 6、17、18 ( 图 1b),空间重建细胞在组织中的位置 19、20 (图 1c ),整合不同分子模态的多组学数据 21 ( 图 1d ),推断细胞与细胞之间的相似性 22 ,跨尺度或视图整合(例如,在形态学和分子分析中) 23 以及缺失模态插补 24 。
The effectiveness of OT comes, however, with drawbacks: because the theory builds on sophisticated mathematics that blends optimization25,26, stochastic differential equation (SDE)11,16 and partial differential equation (PDE)9 and, more recently, deep learning6,7,13,18,23,27, its computations are challenging even by modern machine-learning standards. Developing efficient algorithms for solving OT and its variations, as well as methodologies to make OT applicable to real-world problems, is a significant hurdle for wider adoption28. This serves as the motivation of this Primer for a focused exploration of characteristics and unique potential of OT for single-cell and spatial omics.
然而,OT 在有效的同时也有缺点:因为该理论建立在融合了优化 25、26 、 随机微分方程 (SDE) 11、16 和偏微分方程 (PDE) 9 以及最近的深度学习 6、7、13、18、23、27 的复杂数学基础之上,即使按照现代机器学习标准,其计算也具有挑战性。开发用于解决 OT 及其变体的有效算法,以及使 OT 适用于实际问题的方法是其广泛应用的重大障碍 28。 这成为本入门指南的动机,旨在重点探索 OT 在单细胞和空间组学中的特征和独特潜力。
In this Primer, we introduce the mathematical and computational principles of OT, to guide novel applications. We provide the reader with intuitive explanations of how seemingly unrelated mathematical approaches for analysing single-cell data can be unified through OT theory and how that theory has triggered recent advances in deep learning. We provide an overview of the broad range of biological applications, demonstrating the successes of OT in single-cell biology.
在本入门指南中,我们介绍了 OT 的数学和计算原理,以指导其创新应用。我们为读者提供直观的解释,解释如何通过 OT 理论统一看似毫不相关的单细胞数据分析数学方法,以及该理论如何推动深度学习的最新进展。我们概述了 OT 在生物学领域的广泛应用,并展示了其在单细胞生物学中的成功。
Experimentation 实验
This section introduces the building blocks of OT theory, which we illustrate using representative examples from single-cell and spatial omics.
本节介绍 OT 理论的构成要素,我们使用单细胞和空间组学的代表性示例进行说明。
We first describe the mathematical concept of transport (before turning to the optimal qualifier). In mathematics, transport refers to the various ways to describe the transformation of one point cloud into another. In the simplest setting, these point clouds can describe several particles in the 3D physical space. In our case, such point clouds represent descriptors of high-dimensional data derived from single-cell or spatial omics as vectors in , in which d denotes the dimension of the data, determined by the number of genes or other biological features captured in the measured profile. To introduce the core concepts of OT, we introduce an example from single-cell biology.
我们首先描述迁移的数学概念(然后再转向最优限定词)。在数学中,迁移指的是描述一个点云到另一个点云变换的各种方式。在最简单的情况下,这些点云可以描述三维物理空间中的多个粒子。在我们的例子中,这些点云表示来自单细胞或空间组学的高维数据的描述符,作为 中的向量,其中 d 表示数据的维度,由测量曲线中捕获的基因或其他生物特征的数量决定。为了介绍迁移的核心概念,我们引入一个单细胞生物学的例子。
Example 1: reconstructing the temporal evolution of cell populations
示例 1:重建细胞群体的时间演变
Consider the responses over time of each cell in a (heterogeneous) cell population to a molecular stimulus or perturbation (such as a developmental or environmental signal, gene knockouts or drug treatment)5,6. To study this process, a few thousand cells are sampled from a large population at each of several different time points along a time course and profiled using single-cell RNA sequencing (scRNA-seq). Because of the destructive nature of scRNA-seq, different cells are profiled at each time point. To model this process, each point x represents the recorded features of a single cell from scRNA-seq (Fig. 1a,b). Each feature (dimension) of that point x tracks the expression level of each studied gene in that cell at measurement time. Two consecutive snapshots can be seen as two point clouds or, alternatively, as two tabular data sets X = [x1, …, xn] and Y = [y1, …, ym]: each of the n or m rows contains a cell and its d-dimensional feature representation, in which each column denotes a particular feature, such as the expression level of a gene (Fig. 1a). To understand and reconstruct the temporal evolution of the cell population over time, or how cells in one time point transition to become the cells in a later time point, we aim to provide an informed guess on an alignment or a map that relates the two sets of cells X and Y.
考虑 (异质) 细胞群中每个细胞对分子刺激或扰动 (如发育或环境信号、基因敲除或药物治疗) 随时间的响应 5、6 。 为了研究这一过程,在时间过程中的几个不同时间点从大量细胞中取样数千个细胞,并使用单细胞 RNA 测序 (scRNA-seq) 进行分析。由于 scRNA-seq 的破坏性,每个时间点都会对不同的细胞进行分析。为了模拟这个过程,每个点 x 代表来自 scRNA-seq 的单个细胞的记录特征 (图 1a、b )。该点 x 的每个特征 (维度) 跟踪测量时该细胞中每个研究基因的表达水平。两个连续的快照可以看作两个点云,或者两个表格数据集 X = [ x 1 , ..., x n ] 和 Y = [ y 1 , ..., y m ]:其中 n 或 m 行中的每一行包含一个细胞及其 d 维特征表示,其中每一列表示一个特定特征,例如基因的表达水平(图 1a )。为了理解和重建细胞群随时间的演变,或者一个时间点的细胞如何转变为稍后时间点的细胞,我们旨在对关联两组细胞 X 和 Y 的比对或图谱提供有根据的猜测。
Parameterizing transport 参数化传输
Defining a transport from data set X to Y is equivalent, intuitively, to associating, matching, aligning or mapping each of the elements in X to another in Y, similar to the case of cell profiles recorded at two different points in time resulting in measurement snapshots X and Y. Although there are countless ways to propose such associations, consider first a naive approach that does not usually result in a valid transport: suppose one associates to each point xi in X its closest neighbour in Y according to some distance. Unfortunately, this approach will most often result in an unbalanced association (Fig. 2a), whereby some of the points yj that are close to many points in X (relative to a distance or cost measure c) will be selected repeatedly, whereas those points yj that are far away will not. In that case, cells from the earlier time point that differentiated during the time interval and show alterations in their molecular profile would not be aligned to any progenitor cell in the later time points. The notion of transport requires, intuitively, finding a balanced way in which all points in X are bijectively associated with points in Y.
直观地讲,定义从数据集 X 到 Y 的传输等同于将 X 中的每个元素关联、匹配、对齐或映射到 Y 中的另一个元素,类似于在两个不同时间点记录的细胞概况的情况,从而产生测量快照 X 和 Y 。尽管有无数种方法可以提出这种关联,但首先考虑一种通常不会产生有效传输的简单方法:假设一个人根据某个距离将 X 中的每个点 x 与 Y 中其最近的邻居关联起来。不幸的是,这种方法通常会导致不平衡的关联 (图 2a ),其中一些靠近 X 中的许多点(相对于距离或成本度量 c )的点 y 将被重复选择,而那些距离较远的点 y 则不会。在这种情况下,来自较早时间点的细胞在时间间隔内分化并显示其分子概况发生变化,将不会与后期时间点的任何祖细胞对齐。直观地说,传输的概念要求找到一种平衡的方式,使 X 中的所有点都与 Y 中的点双射关联。
图 2:从最近邻分配到最优运输。

a, Assigning to each point xi (for example, a single cell) its nearest neighbour yj results in unbalanced assignments. b, To enforce a balanced matching, and when the number of points in each set is the same n = m, permutations are used to encode a one-to-one bijective matching. In that plot, σ is an arbitrary permutation, whereas σ⋆ is that with the lowest cost. c, A natural extension for weighted point clouds, with respective weights ai and bj of the source and target distribution, of possibly different sizes n ≠ m is given by transportation plans P, or couplings with suitable marginals. P⋆ refers to the optimal transport plan. d, Moving towards the continuous setting, in which both measures μ and ν have a density, the variable of interest becomes a pushforward map, that is able to reconstruct ν by applying a map T (or the optimal transport map T⋆) to all the points in the support of measure μ. Across all panels, red and blue represent the source and target distributions, respectively.
a 、将每个点 x (例如,单个单元格)分配给其最近邻 y 会导致分配不平衡。 b 、为了强制平衡匹配,并且当每组中的点数相同 n = m 时,使用排列来编码一对一的双射匹配。在该图中, σ 是任意排列,而 σ ⋆ 是成本最低的排列。 c 、加权点云的自然延伸,源和目标分布的权重分别为 a 和 b ,可能大小不同 n ≠ m , 由运输计划 P 或具有合适边际的耦合给出。P ⋆ 指最佳运输计划。d 、 转向连续设置,其中度量 μ 和 ν 都具有密度,感兴趣的变量变成前推图,能够通过将图 T (或最佳运输图 T ⋆ )应用于度量 μ 支持下的所有点来重建 ν 。在所有面板中,红色和蓝色分别代表源分布和目标分布。
One-to-one matchings 一对一匹配
The simplest transport model to go from X to Y can occur when n = m, in which case it can be parameterized as a one-to-one matching. Such matching can be encoded through a permutation σ in the set of permutations of size n, of which there are n!. Intuitively, a permutation is a list (σ1, …, σn), in which each of the n integers from 1 to n appears exactly once, rearranged in any arbitrary order. A permutation is then interpreted as stating that the ith element of X is associated to the σith element in Y, in which the point xi is tied with . Permutations enforce that all points in X are associated with all points in Y and vice versa, using the inverse permutation σ−1 (Fig. 2b), in that each progenitor cell from an earlier time point is mapped to one descendant cell recorded at a later time point or after a perturbation has occurred. At a more conceptual level, permutations are bijections from and to the set {1, …, n}, and there are exactly n! of them.
从 X 到 Y 的最简单的传输模型可能发生在 n = m 时,在这种情况下它可以被参数化为一对一匹配 。这种匹配可以通过在大小为 n 的排列集合 中的排列 σ 进行编码,其中有 n !直观地讲,排列是一个列表 ( σ 1 , ..., σ n ),其中从 1 到 n 的 n 个整数中的每一个都出现一次,以任意顺序重新排列。然后,排列被解释为表明 X 中的第 i 个元素与 Y 中的第 σ 个元素相关联,其中点 x 与 相关联。置换强制 X 中的所有点与 Y 中的所有点相关联,反之亦然,使用逆置换 σ −1 (图 2b ),其中来自较早时间点的每个祖细胞都会映射到在较晚时间点或发生扰动后记录的一个后代细胞。从更概念化的层面来看,置换是从集合 {1, …, n } 到集合 {1, …, n } 的双射,并且恰好有 n ! 个。
Transportation plans 交通计划
Although intuitive and simple, permutations cannot be used if the number of points in X and Y is different, namely, n ≠ m. This limitation is particularly relevant in the context of biological examples, in which various factors contribute to different numbers of points in data sets X and Y. Technically, the discrepancy could be attributed to variations in the number of cells profiled. More fundamentally, biological processes are governed by events such as cell division at different rates along differentiation. Additionally, the nature of the data resembles a fate map rather than a lineage; cells in differentiation processes have multiple non-zero probabilities of transforming into different fates, with only one being realized. This makes the direct application of permutations unsuitable for capturing the intricacies of such biological phenomena and, more generally, to model a transport between weighted point clouds, in which a probability weight ai > 0 (respectively bj) is associated with each point xi in X (respectively yj in Y). A natural generalization for permutations can be found in n × m rectangular coupling matrices P, or transport plan. Each entry Pij describes whether a point xi is matched to point yj. The entry is 0 when there is no association, but, rather than indicating by a binary 1 whether xi is associated to yj, the value in Pij quantifies an association strength, namely, how much of the weight of point xi is transferred to yj (Fig. 2c). When computing a coupling between two snapshots of single-cell measurements, it provides a probabilistic assignment of which progenitor cell would most likely transform into which descendant cell. This is roughly analogous to a cell fate map (but not a lineage map, which is by definition deterministic and is irrelevant here because the measurements are destructive, such that no progenitor cell had any real descendants). To ensure that all masses are conserved, entries of P should be non-negative and such that P1m = a and . The set of admissible matrices is then denoted as
尽管排列直观简单,但如果 X 和 Y 中的点数不同,即 n ≠ m ,则无法使用排列。这种限制在生物学示例中尤其重要,因为各种因素会导致数据集 X 和 Y 中的点数不同。从技术上讲,这种差异可以归因于所分析的细胞数量的变化。更根本的是,生物过程受细胞分裂等事件的支配,这些事件沿着分化过程以不同的速率进行。此外,数据的性质更像是命运图而不是谱系;分化过程中的细胞具有转变为不同命运的多个非零概率,但只有一个能够实现。这使得直接应用排列不适合捕捉此类生物现象的复杂性,更一般地说,不适合模拟加权点云之间的传输,其中概率权重 a > 0(分别为 b )与 X 中的每个点 x (分别为 Y 中的 y )相关联。置换的自然推广可以在 n × m 矩形耦合矩阵 P (或称迁移规划) 中找到。每个元素 P i j 描述点 x 是否与点 y 匹配。 当没有关联时,条目为 0,但是, P i j 中的值不是用二进制 1 表示 x 是否与 y 关联,而是量化关联强度,即点 x 的权重有多少转移到 y (图 2c )。在计算两个单细胞测量快照之间的耦合时,它提供了一个概率分配,即哪个祖细胞最有可能转化为哪个后代细胞。这大致类似于细胞命运图(但不是谱系图,谱系图根据定义是确定性的,并且在这里无关紧要,因为测量是破坏性的,因此没有祖细胞有任何真正的后代)。为了确保所有质量都守恒, P 的条目应该是非负的,并且 P 1 m = a 和 。然后,可接受矩阵集表示为
Pushforward maps 推进地图
Yet another conceptual leap in the OT theory can be achieved by moving from discrete formulations to the continuous regime. Here, point clouds X and Y become intuitively of infinite size and give way to probability measures (Fig. 2d). Now, measurements X and Y are simply realizations of the underlying distribution μ (the distribution over cellular states at one time point) and ν (the distribution of cell states at a later time point). The permutations that were useful to parameterize one-to-one mappings for point clouds of equal size have their counterpart in the more advanced notion of pushforward map, or maps that are such that T♯μ = ν, in which ♯ is the pushforward operator (Box 1). The transport mapT describes, for example, a perturbation effect, as in how an unperturbed population μ responds and evolves into the perturbed population ν = T♯μ (Fig. 1b).
OT 理论的另一个概念飞跃可以通过从离散公式转向连续形式来实现。此时,点云 X 和 Y 直观上变为无限大,并让位于概率测度 (图 2d )。现在,测量值 X 和 Y 仅仅是底层分布 μ (某一时间点的细胞状态分布)和 ν (后续时间点的细胞状态分布)的实现。用于参数化等大小点云一对一映射的置换在更高级的概念 “前推图” 中可以找到对应,即满足 T♯ μ = ν 的映射 ,其中 ♯ 是前推算子(框 1 )。例如, 传输图 T 描述了一种扰动效应,即未受扰动的种群 μ 如何响应并演化为受扰动的种群 ν = T♯ μ (图 1b )。
Evaluating a transport cost
评估运输成本
Through either a permutation, a coupling matrix or a pushforward map, we have defined in the previous section valid ways to transport a (weighted) point cloud or a probability distribution into another predefined configuration. For each of these scenarios, assuming there is a choice of possible maps, what would constitute a good or efficient transport is determined. Here, a good or efficient transport is one that proposes a meaningful alignment between different measurements.
在上一节中,我们定义了通过置换、耦合矩阵或前推映射,将(加权)点云或概率分布传输到另一个预定义配置的有效方法。对于每种情况,假设存在多种可能的映射,则需要确定一种良好或高效的传输方式。在这里,良好或高效的传输是指能够在不同测量结果之间建立有意义的对齐的传输方式。
To define a notion of efficiency, we rely on a cost function c between a pair of points to derive various objective functions, one for each of the ways we have defined transport. Concretely, we need to select a cost function for which, based on their molecular features, cells are aligned to their most likely cell state in the subsequent measurement. Although one can use Euclidean distances for low-dimensional data or cosine29 or correlation-based distances30 for RNA-seq data, robust choices of cost metrics are active areas of research31,32,33,34.
为了定义效率的概念,我们依赖一对点之间的成本函数 c 来推导各种目标函数,每种目标函数对应我们定义的传输方式。具体来说,我们需要选择一个成本函数,根据细胞的分子特征,将细胞与其在后续测量中最可能的细胞状态对齐。虽然低维数据可以使用欧氏距离,RNA 测序数据可以使用余弦距离 29 或基于相关性的距离 30 ,但稳健的成本指标选择仍然是当前研究的热点 31、32、33、34 。
In the case of permutations, a natural global cost computed from local costs between pairs of matched points can be defined by comparing matched points, as
对于排列,可以通过比较匹配点来定义由匹配点对之间的局部成本计算出的自然全局成本 ,如下所示
The natural extension of this idea when transporting mass between weighted point clouds yields a sum of costs between pairs, weighted by the amount transferred between these two points:
当在加权点云之间传输质量时,这个想法的自然延伸会产生对之间的成本总和,该总和由这两点之间传输的量加权:
The resulting cost can be interpreted as a quantification, or distributional distance, between the two point clouds μ and ν. Besides the plan P itself, the OT distance:
最终的成本可以解释为两个点云 μ 和 ν 之间的量化或分布距离。除了规划 P 本身之外,OT 距离:
is an important quantity used in the analysis of single-cell or spatial omics. It is also often employed as a loss function in machine-learning applications to quantify how close the output distribution of a model resembles the data set used to train the model.
是单细胞或空间组学分析中的一个重要参数。它也常被用作机器学习应用中的损失函数,以量化模型输出分布与用于训练模型的数据集的相似程度。
A natural objective for a pushforward transport map T from to is given by the integral:
从 到 的前推传输图 T 的自然目标由积分给出:
That integral blends elements from both costs mentioned earlier, borrowing from equation (2) the idea of comparing each point x with the point T(x) it is mapped to, while also incorporating mass considerations as that cost is weighted by the mass μ at x. Equation (5) represents the famous Monge35 formulation of OT.
该积分融合了前面提到的两种成本的元素,借鉴了方程 ( 2 ) 中将每个点 x 与其映射到的点 T ( x ) 进行比较的思想,同时还融入了质量的考虑因素,因为该成本由 x 处的质量 μ 加权。方程 ( 5 ) 代表了著名的 Monge 35 OT 公式。
Although using the same notation for all three formulas mentioned earlier might be seen as a slight abuse of notation, we do so because this highlights the unifying idea of summing — with either single-indexed or double-indexed sums, or with integrals — granular contributions brought by costs computed between pairs of points.
尽管对前面提到的所有三个公式使用相同的符号 可能被视为对符号的轻微滥用,但我们这样做是因为这突出了求和的统一思想——使用单指标或双指标总和,或使用积分——由点对之间计算的成本带来的细粒度贡献。
Finding an optimal transport
寻找最佳运输方式
Computational OT is the field concerned with efficiently finding a transport, which can either be a permutation σ, a coupling matrix P or a map T, that has a low cost . In each of these three cases, minimizing results in a constrained optimization problem. These problems are, respectively, known in the literature as the optimal assignment, the Kantorovich36 and the Monge35 problems. This search creates a host of challenges that we briefly survey. We first describe exact methods, which aim to probably find the best possible transport. Because of the computational challenges faced by these methods, we next introduce various approaches that rely instead on regularization and/or neural network parameterizations to obtain approximate yet tractable solutions.
计算 OT 是研究如何高效地找到一种传输方式的领域,这种传输方式可以是排列 σ 、耦合矩阵 P 或映射 T ,且具有较低的成本 。在这三种情况下,最小化 都会导致一个受约束的优化问题 。在文献中,这些问题分别被称为最优分配、Kantorovich 36 问题和 Monge 35 问题。这种探索带来了一系列挑战,我们将对其进行简要概述。我们首先描述精确的方法,旨在找到最佳的传输方式。由于这些方法面临的计算挑战,接下来我们将介绍各种依赖于正则化和/或神经网络参数化的方法来获得近似但易于处理的解决方案。
Solving optimal transport exactly
精确求解最优传输
Finding a permutation σ that minimizes equation (2) or a coupling P for equation (3) is the seminal optimization problem, appearing as early as the first half of the twentieth century. The former can be solved with the Hungarian algorithm37, with a worst-case complexity scaling as O(n3). The latter is widely recognized as a central piece of optimization theory, which provided the impetus for the entire field of linear programming38. The problem of finding an optimal coupling P was formulated by Hitchcock39, Kantorovich36 and Koopmans40 (Box 2). Most computational approaches solving it rely on variants of the network simplex41 or the auction algorithm42, with computational cost . That cubic cost is often prohibitive for large-scale applications. In addition, these algorithms can be difficult to implement on parallel architectures, such as GPUs, because they involve sequential discrete selections, such as the pivot rule in the simplex.
寻找使方程 ( 2 ) 最小化的排列 σ 或方程 ( 3 ) 的耦合 P 是一个开创性的优化问题,早在二十世纪上半叶就出现了。前者可以用匈牙利算法 37 求解,最坏情况复杂度为 O ( n3 )。后者被广泛认为是最优化理论的核心部分,它为整个线性规划领域 38 提供了推动力。寻找最优耦合 P 的问题由 Hitchcock 39 、Kantorovich 36 和 Koopmans 40 (框 2 )提出。大多数解决该问题的计算方法依赖于网络单纯形 41 或拍卖算法 42 的变体,计算成本为 。对于大规模应用来说,这种立方成本通常是过高的。此外,这些算法很难在并行架构(如 GPU)上实现,因为它们涉及顺序离散选择,例如单纯形中的枢轴规则。
Because it requires optimizing over functions, while handling a non-convex pushforward constraint, equation (5) is much harder to solve in practice. Given two probability measures μ and ν, the feasible set of maps that can push forward μ to ν is not convex, making the toolbox of convex optimization irrelevant. In the common case in which the cost between two points is the squared-Euclidean distance, c(x, y) = ∥x − y∥2, two approaches have been proposed. The first approach, proposed by Benamou and Brenier43, re-parameterizes the OT problem by introducing a discrete sequence μk of measures, for 0 ≤k ≤T, that interpolates, using the convention μ0 ≔ μ and μT ≔ ν, between the two measures to be compared. Using another discretization (in space), this method proposes to parameterize OT using the continuity equation (13) and advection of velocity fields vt. Its main innovation is to re-parameterize the problem of minimizing the total kinetic energy needed to realize that sequence (itself equal to the total Euclidean cost) as a function of (μtvt, μt) rather than (μt, vt) to recover a convex problem. This requires, however, a discretization not only in time t but also in space, which is only tractable for low-dimensional problems. The second approach relies on exploiting Brenier’s theorem44 (Box 3) to reframe the OT problem as a PDE problem, known as the Monge–Ampère equation3. Because both approaches require a grid discretization on the space of observations, they can only be implemented when observations are low-dimensional, making them unsuitable for the high-dimensional problems of single-cell and spatial omics. However, as we show in the next section, Brenier’s theorem44 (Box 3) does play an important role in neural network-inspired approaches as well as in dynamic formulations of OT.
由于需要对函数进行优化,同时处理非凸的前推约束,方程 ( 5 ) 在实践中更难求解。给定两个概率测度 μ 和 ν ,可以将 μ 前推到 ν 的可行映射集不是凸的,因此凸优化工具箱变得无关紧要。在两点之间的成本是平方欧几里得距离的常见情况下, c ( x , y ) = ∥ x − y ∥ 2 ,已提出了两种方法。第一种方法由 Benamou 和 Brenier 43 提出,通过引入一个离散的测度序列 μ k (其中 0 ≤ k ≤ T )重新参数化 OT 问题,使用约定 μ 0 ≔ μ 和 μ T ≔ ν 在两个要比较的测度之间进行插值。该方法采用另一种空间离散化方法,提出使用连续性方程 ( 13 ) 和速度场平流 v t 来参数化 OT。其主要创新之处在于,将最小化实现该序列所需的总动能(本身等于总欧氏成本)的问题重新参数化为 ( μ t v t , μ t ) 的函数,而不是 ( μ t , v t ) 的函数,从而恢复凸问题。 然而,这不仅需要在时间 t 上离散化,还需要在空间上离散化,而这只适用于低维问题。第二种方法依赖于利用 Brenier 定理 44 (框 3 )将 OT 问题重新定义为 PDE 问题,即 Monge-Ampère 方程 3 。由于这两种方法都需要在观测空间上进行网格离散化,因此它们只能在观测值为低维时实现,这使得它们不适用于单细胞和空间组学的高维问题。然而,正如我们在下一节中所示,Brenier 定理 44 (框 3 )在神经网络启发方法以及 OT 的动态公式中确实发挥着重要作用。
Data-driven optimal transport solvers
数据驱动的最优传输求解器
Equations (2), (3) and (5) provide an intuitive formalism to solving OT problems, but yield intractable computations when used in practice with large sample sizes (n ≥ 103) or high-dimensional (d ≫ 3) data, both settings being the working assumption of single-cell and spatial omics. This has led to several proposals, focusing on the Kantorovich and Monge problems, to compute efficiently an n × m coupling matrix between samples, or a map .
方程 ( 2 )、( 3 ) 和 ( 5 ) 为解决 OT 问题提供了一种直观的形式化方法,但在实际处理大样本量( n ≥ 103 )或高维数据( d≫3 )时,会产生难以处理的计算,而这两种情况都是单细胞和空间组学的工作假设。这导致了多项提案的提出,这些提案主要针对 Kantorovich 问题和 Monge 问题,旨在高效地计算样本之间的 n × m 耦合矩阵,或映射 。
Solvers to compute coupling matrices
计算耦合矩阵的求解器
Although linear programme solvers can be used to solve equation (3), they scale poorly as n grows. Instead, most solvers currently in use output an approximate solution using penalized approaches. Among those, entropic regularization25 is arguably the most popular approach, because it relies on the Sinkhorn algorithm, a fixed-point iteration that only uses matrix–vector products (Box 4). This algorithm can also yield estimators for the OT map T(5), as presented in the next section.
虽然线性规划求解器可以用来求解方程 ( 3 ),但它们的扩展性随着 n 的增长而变差。目前使用的大多数求解器都使用惩罚方法输出近似解。其中,熵正则化 25 可以说是最流行的方法,因为它依赖于 Sinkhorn 算法,这是一种仅使用矩阵向量积的定点迭代算法(框 4 )。该算法还可以为 OT 映射 T ( 5 ) 提供估计量,如下一节所述。
A more recent strand of solvers relies on low-rank approximations of both cost and coupling matrices45,46, by parameterizing variable P as the product of three matrices QD(1/g)RT of respective sizes n × r, r × r and r × m. Although harder to implement than the Sinkhorn algorithm, these solvers have the favourable property that their runtime becomes linear in the size of point clouds, under the assumptions that the rank d of cost matrices is small compared with sample sizes and the restriction that only couplings of rank r are considered. Although the former assumption is often observed as the rank of a pairwise squared-Euclidean distance matrix between n and m points in is at most d + 2, restricting optimization to couplings of rank r induces solutions that intuitively restrict the displacement of mass between two point clouds to move through r intermediary hubs46, which might recover hierarchical structures such as cell types present in the aligned data47.
较新的一类求解器依赖于成本矩阵和耦合矩阵的低秩近似 45、46 ,通过将变量 P 参数化为三个矩阵 QD (1/ g ) RT 的乘积 , 其大小分别为 n × r 、 r × r 和 r × m 。虽然比 Sinkhorn 算法更难实现, 但它们具有良好的特性,即在成本矩阵的秩 d 小于样本大小的假设以及仅考虑秩为 r 的耦合的限制下,它们的运行时间与点云的大小呈线性关系。尽管前一种假设通常被观察到为 中 n 和 m 点之间的成对平方欧几里得距离矩阵的秩最多为 d + 2,但将优化限制为秩为 r 的耦合会诱导直观地限制两个点云之间的质量位移通过 r 个中间中心 46 的解决方案,这可能会恢复对齐数据 47 中存在的层次结构(例如细胞类型)。
Solvers to compute transport maps
用于计算传输图的求解器
The exact and data-driven solvers to compute coupling matrices we described earlier cannot operate on unseen samples: they only return an alignment or coupling of those data points initially considered in the computation. Conversely, if we, for example, wish to infer the descendants of an unseen sample or data set, or predict the effect of a drug on new cells from a different patient, we need solvers that act out-of-sample. In addition to that computational shortcoming, the solvers mentioned earlier have a statistical flaw, as approaches based on discrete samples, such as equation (3), tend to overfit data, leveraging information from finite samples to the extent that such couplings do not generalize well to new points48 because of the curse of dimensionality49.
我们之前描述的用于计算耦合矩阵的精确和数据驱动的求解器无法对未见过的样本进行操作:它们仅返回计算中最初考虑的数据点的对齐或耦合。相反,如果我们希望推断未见过的样本或数据集的后代,或者预测药物对不同患者的新细胞的影响,则需要样本外求解器。除了计算上的缺陷之外,前面提到的求解器还存在统计缺陷,因为基于离散样本的方法(例如公式 ( 3 ))往往会过度拟合数据,利用有限样本中的信息,以至于由于维数灾难 49 ,这种耦合不能很好地推广到新点 48 。
Concretely, when looking for a map T that solves equation (5) for a pair of measures μ, ν and a cost function c, the challenge is to work out, from samples x1, …, xn ~ μ and y1, …, ym ~ ν, a function that provides a plausible substitute for T⋆.
具体来说,当寻找一个映射 T 来求解方程 ( 5 ) 中一对测度 μ 、 ν 和一个成本函数 c 时 ,挑战在于从样本 x 1 , ..., x n ~ μ 和 y 1 , ..., y m ~ ν 中找出一个函数 来为 T ⋆ 提供合理的替代品。
The benefit of such an approach is to recover a function that can generalize to new points, rather than just obtain a matching matrix between existing samples. In that context, two main approaches stand out.
这种方法的好处是恢复一个可以推广到新点的函数,而不仅仅是获得现有样本之间的匹配矩阵。在这方面,有两种主要方法脱颖而出。
The first approach extends the estimates produced by Sinkhorn solvers (Box 4) out-of-sample, owing to duality2 (Box 2). In brief, using point clouds (x1, …, xn) and (y1, …, ym), compared through cost function c, this approach consists in solving first equation (19) to recover the two dual variables that are fixed points of equation (21). These two vectors α⋆, β⋆ contain n + m values, one for each of the x and y points contained in the source (x1, …, xn) and target (y1, …, ym) distributions. The following formulas31,50,51 can be used to extend these values to out-of-sample points:
第一种方法将 Sinkhorn 求解器(框 4 )产生的估计值扩展到样本外, 这是由于对偶性 2 (框 2 )造成的。简而言之,使用点云( x1 ,..., xn )和 ( y1 , ...,ym ) ,通过成本函数 c 进行比较,该方法包括求解第一个方程( 19 )以恢复两个对偶变量 , 它们是方程( 21 ) 的不动点。这两个向量 α⋆ , β⋆ 包含 n + m 个值,源( x1 ,..., xn ) 和目标( y1 ,..., ym ) 分布中包含的每个 x 和 y 点都有一个值 。 以下公式 31、50、51 可用于将这些值扩展到样本外点:
which can then be plugged into a generalized Brenier-type formula52, to recover in full generality
然后可以将其代入广义的 Brenier 型公式 52 中,以完全恢复一般性
This formula requires invertibility concerning the second variable, of the map ∇1c(x, ⋅) at any x, a condition often referred to as the twist condition. This formula is of course notably simpler for common costs c. For instance, when the cost is the squared Euclidean cost, this recovers
该公式要求映射 ∇ 1 c ( x , ⋅ ) 的第二个变量在任意 x 处具有可逆性,这一条件通常被称为扭转条件 。当然,对于常见的成本 c 来说,这个公式要简单得多。例如,当成本是欧几里得成本的平方时,公式可以恢复
in which can be interpreted as a discrete Gibbs distribution, depending on x, using values and temperature ε.
其中 可解释为离散吉布斯分布,取决于 x ,使用值 和温度 ε 。
The second approach is based on neural networks. Finding a function T that approximately maps a distribution onto another, that is, T♯μ = ν, using samples from both measures is a fundamental task in machine learning that is often handled using neural networks. Here, approaches diverge based on two different scenarios. In the supervised setup, in which paired samples from a coupling for μ, ν are given, that is, input–output pairs (xi, yi), estimating such maps T requires minimizing an empirical reconstruction loss, as in ℓ(yi, T(xi)). OT methods tackle, on the contrary, the more ambitious and somewhat only partially supervised setting, in which unpaired data sets and are given. Such problems have been described in the past as generative modelling problems53,54, or alternatively as normalizing flows and variants55, notably when either of these measures is simple to sample from. In that sense, OT provides some novelty. From a descriptive perspective, OT maps do more than simply push forward a measure onto another; they should in principle be the best of such maps. And, it is by asking that extra requirement that we can get, in exchange, a useful inductive bias to guide the selection of these maps. That bias is given more precisely by Brenier’s theorem44, which has led refs. 56,57 to parameterize OT maps as gradients of neural networks6,9,58,59, or directly as a vector-valued neural network7 using a regularizer.
第二种方法基于神经网络。使用来自两个度量的样本,找到一个将一个分布近似映射到另一个分布的函数 T ,即 T♯ μ = ν ,是机器学习中的基本任务,通常使用神经网络来处理。在这里,方法根据两种不同的情况而有所不同。在监督设置中,给定来自 μ , ν 的耦合的配对样本,即输入输出对 ( x , y ),估计这样的映射 T 需要最小化经验重建损失,如 ℓ ( y , T ( x ) )。相反,OT 方法处理的是更具挑战性且仅部分监督的设置,其中给定未配对的数据集 和 。过去,此类问题被描述为生成建模问题 53、54 ,或者称为规范化流和变体 55 ,特别是当其中任何一个度量都易于采样时。从这个意义上说,OT 提供了一些新颖之处。从描述性的角度来看,OT 映射不仅仅是简单地将一个测度推到另一个测度上;原则上,它们应该是此类映射中最好的。而且,正是通过提出这个额外的要求,我们才能获得一个有用的归纳偏差来指导这些映射的选择。这种偏差由 Brenier 定理 44 更精确地给出,该定理引发了文献[2]。 56、57 将 OT 图参数化为神经网络 6、9、58、59 的梯度 ,或直接使用正则化器将其参数化为向量值神经网络 7 。
Extensions of optimal transport
最优传输的扩展
So far, we have considered standard formulations of OT, illustrated using the example of modelling cell differentiation into various cell lineages or how cell populations respond to perturbations. However, several key characteristics of biological systems require adaptations of classical OT, including allowing for cell division, migration and death, integrating different data modalities or tracking cellular responses continuously in time. In the following, we introduce extensions of OT that can capture these characteristics.
到目前为止,我们已经探讨了场理论(OT)的标准公式,并以模拟细胞分化成各种细胞谱系或细胞群体如何响应扰动为例进行了说明。然而,生物系统的几个关键特性需要对经典场理论进行调整,包括允许细胞分裂、迁移和死亡,整合不同的数据模式,或持续跟踪细胞随时间的变化。接下来,我们将介绍能够捕捉这些特性的场理论的扩展。
Partial matchings 部分匹配
The conservation of mass principle is fundamental to all definitions of OT mentioned earlier and distinguishes it from simpler nearest-neighbour-based matching approaches or from attention mechanisms in transformers60. It is, however, possible to escape that binary view and introduce a gradual approach to parameterize the degree to which one expects a coupling P or a map T to obey that constraint. This provides greater flexibility when modelling cellular dynamics that are subject to birth (from division or migration) and death events5,18,61,62 or data sets that contain different numbers of measurements21.
质量守恒原理是前面提到的所有 OT 定义的基础,并将其与更简单的基于最近邻的匹配方法或 Transformer 中的注意机制 60 区分开来。然而,可以摆脱这种二元视图,并引入一种渐进的方法来参数化人们期望耦合 P 或映射 T 遵守该约束的程度。这在模拟受出生(分裂或迁移)和死亡事件 5、18、61、62 或包含不同测量次数 21 的数据集影响的细胞动力学时提供了更大的灵活性。
The key insight in such approaches is to relax and dualize such mass conservation laws (Box 2). In the case of couplings (equation (3)), this can be achieved by dropping the feasible set (1) and adding to the objective a multiple of Δ(P1m∣a) and Δ(PT1n∣b), in which Δ is a discrepancy function quantifying the difference between two unnormalized distributions63,64. A notable case is given when Δ is the Kullback–Leibler divergence, because of its natural connections with the entropic regularization presented in equation (19). Indeed, rewriting the objective in equation (19) as a Kullback–Leibler divergence itself, one can define the unbalanced entropic transport objective as
此类方法的关键在于放宽并二元化此类质量守恒定律(框 2 )。对于耦合(方程 ( 3 )),可以通过删除可行集 ( 1 ) 并在目标函数中添加 Δ ( P1m∣a ) 和 Δ( P1n∣b ) 的倍数来实现,其中 Δ 是量化两个非正则化分布之间差异的差异函数 63、64 。 一个值得注意的例子是 Δ 是 Kullback-Leibler 散度,因为它与方程 ( 19 ) 中提出的熵正则化有着天然的联系。事实上,将方程 ( 19 ) 中的目标函数重写为 Kullback-Leibler 散度本身,就可以将不平衡熵传输目标定义为
which can be solved using a minor modification of the Sinkhorn algorithm, in which the updates in equation (20) have an extra element-wise exponential operation,
这可以使用对 Sinkhorn 算法稍加修改来求解,其中方程 ( 20 ) 中的更新具有额外的逐元素指数运算,
and, analogously, the log-space updates in equation (21) are simply multiplied by and , respectively.
类似地,方程 ( 21 ) 中的对数空间更新分别简单地乘以 和 。
Example 2: integrating multimodal data
示例 2:整合多模态数据
The increasing emergence of different omic technologies allows researchers to integrate different types of data sources to gain a more comprehensive understanding of cellular processes. These data sources can include, for example, gene expression profiles, DNA methylation profiles, protein–protein interaction data or spatial information at the single-cell level. For instance, consider a study that aims to integrate gene expression profiles X with epigenetic profiles (such as DNA methylation) Y to explore the relationship between gene regulation and epigenetics. Here, each modality lies in a different space and , for example, we are provided with gene expression data and epigenetic data , and an alignment between measurements of different data modalities, in that heterogeneous or incomparable spaces, is required. OT can be used to align the distributions of gene expression levels and DNA methylation patterns across multiple cell types or conditions. This alignment allows for a systematic comparison and identification of genes that show coordinated changes in expression and DNA methylation, shedding light on the regulatory mechanisms underlying cellular processes.
不同组学技术的不断涌现使研究人员能够整合不同类型的数据源,以更全面地了解细胞过程。这些数据源可以包括例如基因表达谱、DNA 甲基化谱、蛋白质 - 蛋白质相互作用数据或单细胞水平的空间信息。例如,考虑一项旨在整合基因表达谱 X 与表观遗传谱(如 DNA 甲基化) Y 以探索基因调控和表观遗传学之间关系的研究。在这里,每种模态位于不同的空间 和 ,例如,我们提供基因表达数据 和表观遗传数据 ,并且需要在异构或不可比的空间中对不同数据模态的测量进行对齐。OT 可用于对齐多种细胞类型或条件下的基因表达水平和 DNA 甲基化模式的分布。这种比对可以系统地比较和识别表现出表达和 DNA 甲基化协调变化的基因,从而揭示细胞过程背后的调控机制。
Multimodal alignments 多模态比对
In all the descriptions of transport given so far, we have relied on the knowledge of a cost function that can quantify the difference between two observations living in . Yet, in many applications, practitioners may wish to align or match data across heterogeneous measurement spaces and , as demonstrated by Example 2. These settings arise, for example, when integrating several modalities (Fig. 1d) or when spatially reconstructing tissues from (partially) non-spatially resolved data (Example 3, Fig. 1c). When operating across different measurement technologies or data spaces, no obvious cost for such heterogeneous observations is known a priori. We thus need to design a new cost objective function for couplings P that can still be used, assuming we have at least two meaningful cost functions and for each data space. The inspiration for this approach lies in the quadratic assignment problem, which now seeks isometric matchings, such that if the mass of a point xi is mostly transported to yj (Pij) and similarly from to (), then the gap between costs and is small (Fig. 3a). Thus, we are aligning two data modalities based on matching the overall sample structure or geometry of the measurements. This principle (Fig. 3a) translates into the following cost65:
到目前为止,在对传输的所有描述中,我们都依赖于成本函数的知识,该函数可以量化位于 中的两个观测值之间的差异。然而,在许多应用中,从业者可能希望跨异构测量空间 和 对齐或匹配数据,如示例 2 所示。例如,在整合几种模态(图 1d )或从(部分)非空间分辨数据空间重建组织(示例 3,图 1c )时,就会出现这些设置。当跨不同的测量技术或数据空间操作时,对于这种异构观测值 没有明显的成本是先验已知的。因此,我们需要为耦合 P 设计一个仍然可以使用的新成本目标函数,假设每个数据空间至少有两个有意义的成本函数 和 。这种方法的灵感来自于二次分配问题,该问题现在寻求等距匹配,使得如果点 x 的质量大部分传输到 y ( P i j ),并且类似地从 传输到 ( ),则成本 和 之间的差距很小(图 3a )。因此,我们根据匹配整体样本结构或测量的几何形状来对齐两种数据模态。该原理(图 3a )转化为以下成本 65 :
and the resulting optimization problem that identifies the optimal P given objective (12) is also known as Gromov–Wasserstein. Although it may seem that evaluating this cost may have a prohibitive O(n2m2) complexity owing to this quadruple sum, the properties of P ensure that this is O(nm(n + m)) in all cases66, and even a far more favourable if both cost matrices and have rank and , respectively67. This quadratic regime can be paired with the Sinkhorn algorithm to yield efficient solvers that are guaranteed to converge to a local optimum. Note that the low-rank approach in ref. 67 goes one step further and results in an overall linear complexity with respect to sample size, which has served as the computational foundation for a few recent applications of Gromov–Wasserstein68.
并由此产生的优化问题,即在给定目标 ( 12 ) 的情况下确定最优 P , 也称为 Gromov – Wasserstein 。尽管由于这个四重和,评估这个成本似乎可能具有令人望而却步的 O ( n2m2 ) 复杂度, 但 P 的属性确保在所有情况下这都是 O ( nm ( n + m )) 66 ,并且如果成本矩阵 和 分别具有秩 和 ,则甚至是更有利的 67。 这种二次方案可以与 Sinkhorn 算法配对,以产生保证收敛到局部最优的高效求解器。请注意,参考文献中的低秩方法。 67 更进一步,得到了关于样本大小的总体线性 复杂度,这为 Gromov–Wasserstein 68 的一些近期应用提供了计算基础。
图 3:最佳运输向多式联运设置和动态公式的扩展。

a, When computing alignments across heterogeneous or incomparable spaces (red) and (blue), the optimal transport (OT) alignment is computed based on matching the overall geometry, or intra-space distances, here measured with two different distance functions d and between two sets of points [x1, x2, x3, …] and [y1, y2, y3, …]. The resulting Gromov–Wasserstein formulation can then be used to provide a correspondence between cells measured through different modalities, for example, single-cell RNA sequencing (RNA-seq) in source distribution μ (red) and ATAC-seq in target distribution ν (blue). b, OT can describe continuous-time dynamics of single cells. The dynamic OT formulation thereby finds the minimal path μt according to an underlying time-varying vector field v(t, ⋅) between distribution μ0 at time t = 0 (dark blue) and μ1 at time t = 1 (light blue). In connection to Brenier’s theorem, continuous-time dynamics of cell populations can be reconstructed along the gradient of the potential function f, that is, ∇f.
a 、在计算异构或不可比空间 (红色)和 (蓝色)之间的比对时,最佳传输 (OT) 比对是根据整体几何或空间内距离的匹配来计算的,这里用两个不同的距离函数 d 和 在两组点 [ x 1 , x 2 , x 3 , …] 和 [ y 1 , y 2 , y 3 , …] 之间进行测量。得到的 Gromov-Wasserstein 公式可用于提供通过不同模态测量的细胞之间的对应关系,例如,源分布 μ (红色)中的单细胞 RNA 测序 (RNA-seq) 和目标分布 ν (蓝色)中的 ATAC-seq。b、 OT 可以描述单细胞的连续时间动态。因此,动态 OT 公式根据底层时变矢量场 v ( t , ⋅ ),找到时间 t = 0 时的分布 μ0 (深蓝色)与时间 t = 1 时的分布 μ1 (浅蓝色)之间的最小路径 μt 。 结合 Brenier 定理,可以沿着势函数 f 的梯度重建细胞群体的连续时间动力学,即 ∇f 。
Dynamic formulations 动态配方
So far, we have considered static OT schemes that map a distribution μ into distribution ν. Biological processes, however, are dynamic: after a signal or perturbation k, cell states evolve gradually over time. Capturing and modelling this temporal continuity is crucial to understanding biological processes. With the growing availability and reduced costs of single-cell omics, it is possible to profile a large number of cells from an evolving cell population μt, along multiple time points, from μ0 at time t = 0 to μ1 at t = 1 (refs. 9,12,13).
到目前为止,我们已经考虑了将分布 μ 映射到分布 ν 的静态 OT 方案。然而,生物过程是动态的:在信号或扰动 k 之后,细胞状态会随时间逐渐演变。捕捉和建模这种时间连续性对于理解生物过程至关重要。随着单细胞组学的普及和成本的降低,我们有可能对一个不断发展的细胞群体 μ t 中的大量细胞进行分析,这些细胞沿着多个时间点,从时间 t = 0 时的 μ 0 到 t = 1 时的 μ 1 进行分析(参考文献 9、12、13 ) 。
As posited by Benamou and Brenier43, the dynamic formulation is ‘already implicitly contained in the original problem addressed by Monge’35 (equation (5)), in which ‘eliminating the time variable was just a clever way of reducing the dimension of the problem’43. When reintroducing time to the OT problem, the transport map becomes a time-dependent flow capable of describing the evolution of a population over time. The Brenier theorem (Box 3) forms a critical bridge that connects the static and dynamic formulation. When considering the squared Euclidean cost and , the OT problem coincides with finding the minimal path or, more concretely, a curve in the space of distributions, minimizing a total length (Fig. 3b). Such path μt can be described through a time-varying vector field v(t, ⋅) which moves particles around, satisfying the continuity equation in fluid dynamics or conservation of mass formula:
正如 Benamou 和 Brenier 43 所假设的,动态公式“已经隐含在 Monge 处理的原始问题中” 35 (方程( 5 )),其中“消除时间变量只是降低问题维度的一种巧妙方法” 43。 当将时间重新引入 OT 问题时,传输图变成了与时间相关的流 ,能够描述种群随时间的演变。Brenier 定理(框 3 )构成了连接静态和动态公式的关键桥梁。当考虑平方欧几里得成本 和 时 ,OT 问题与寻找最小路径 相一致,或者更具体地说,在分布空间中寻找一条曲线,以最小化总长度(图 3b )。这样的路径 μ t 可以通过随时间变化的矢量场 v ( t , ⋅ ) 来描述,该矢量场移动粒子,满足流体动力学中的连续性方程或质量守恒公式:
in which the vector field v(t, ⋅) denotes the speed and μtv(t, ⋅) = Jt corresponds to the momentum. Every curve μt describing the evolution of the measure over time can be interpreted as the fluid flow along a family of vector fields. We are searching for the vector field v(t, ⋅) that satisfies the conservation of mass (13) and minimizes the kinetic energy of the path. The infinitesimal length of such a vector field can be computed via
其中矢量场 v ( t , ⋅ ) 表示速度, μ t v ( t , ⋅ ) = J t 表示动量。每条描述测量值随时间演变的曲线 μ t 都可以解释为流体沿一族矢量场的流动。我们正在寻找满足质量守恒( 13 )且最小化路径动能的矢量场 v ( t , ⋅ )。此类矢量场的无穷小长度可以通过以下公式计算
resulting in the dynamic reformulation of the OT problem
导致 OT 问题的动态重构
This provides an intuition on how OT allows us to study dynamical systems and model population dynamics that follow some optimality criterion (13) through the system of ordinary differential equations (15). Subsequently, a series of dynamic OT methods have been developed9,11,12,13,69, which we explore in the Applications section and discuss their potential in the Outlook section.
这为我们提供了一种直观的理解,即 OT 如何使我们能够通过常微分方程组( 15 )来研究遵循某些最优准则( 13 )的动态系统和模型人口动态。随后,一系列动态 OT 方法被开发出来 9 、 11 、 12 、 13 、 69 ,我们将在“应用”部分进行探讨,并在“展望”部分讨论它们的潜力。
Results 结果
The framework of OT notion of distance, transport plans and transport maps can be used to model key questions of cell and tissue biology. In this section, we describe how to use the distance for interpretation and quantification tasks, how to use the OT plan to align cell populations in space and time and how to use the OT map for making predictions on unobserved samples such as new cell types or patients.
OT 概念的距离、传输计划和传输图谱框架可用于模拟细胞和组织生物学的关键问题。在本节中,我们将描述如何使用距离进行解释和量化任务,如何使用 OT 计划在空间和时间上对齐细胞群体,以及如何使用 OT 图谱对未观察的样本(例如新的细胞类型或患者)进行预测。
OT distance for cell states and niches
细胞状态和生态位的 OT 距离
Many questions in single-cell omics focus on the changes in the composition of cell populations and their molecular heterogeneity in space and time. Measuring and identifying different cellular states or environments, however, requires a meaningful notion of metric — a challenging task within single-cell genomics and thus an area of active research70. By providing a well-founded, geometrically driven approach to computing a matching between unaligned point clouds, OT induces a theoretically well-characterized distance between distributions or populations, with multiple use cases across single-cell and spatial analysis frameworks.
单细胞组学中的许多问题集中在细胞群体组成的变化及其在空间和时间上的分子异质性上。然而,测量和识别不同的细胞状态或环境需要一个有意义的度量概念——这在单细胞基因组学中是一项具有挑战性的任务,因此也是一个活跃的研究领域 70 。通过提供一种有理有据的、几何驱动的方法来计算未对齐点云之间的匹配,OT 可以推导出分布或群体之间理论上特征明确的距离,并在单细胞和空间分析框架中有多种用例。
Single-cell omics 单细胞组学
Genetic and chemical perturbations can profoundly affect the cellular phenotype. It is now possible to perform large-scale screens where cells are perturbed by different genetic or chemical perturbations and then profiled at the single-cell level. Such pooled Perturb-seq screens71 have been used to identify gene function by gene knockout or activation screens72 and categorize coding and non-coding variants into distinct levels of perturbation impact. Furthermore, increasingly large scRNA-seq screens become available that allow assessing the effects of small-molecule drugs73. A first generation of analyses thereby quantified the overall perturbation effect by measuring average cellular responses at the gene71 or cell level74. The outcome of different perturbations might, however, strongly vary between heterogeneous cell states within a population, such as cellular behaviours not captured through modelling averages. By comparing the unperturbed and perturbed cell population through comparing their distributions, OT can capture such fine-grained but heterogeneous responses. To capture the magnitude of the effect of a perturbation k on a cell population, we can compute the OT distance OT(X, Yk) between a data sample of unperturbed cells X and perturbed cells Yk. By aligning and then summing the difference between aligned cell states, OT quantifies the strength of heterogeneous cellular responses (Fig. 4a). Building upon this intuition, Bunne et al.6 analyse the strength of different cancer drugs based on scRNA-seq profiles of two melanoma cell lines conducted through 4i multiplexing75 (Fig. 4b,c). Examining the OT cost of different cancer drug treatments for different cell states as well as the OT cost summed over all cell states demonstrates how the OT distance can serve as a measure for identifying drug sensitivities of distinct cellular states to different drugs. Apoptosis inducers (staurosporine), proteasome inhibitors (ixazomig and carfilzomib or the combination treatment carfilzomib + pomalidomide + dexamethasone), microtubule-stabilizing agents (paclitaxel) and ATP competitors for multiple tyrosine kinases such as KIT and BCR–ABL (dasatinib) induced substantial feature changes in all cellular states and thus showed high transport costs (Fig. 4c). Finally, the OT distance can also be utilized to create a map of the patient-state space that highlights sources of patient-to-patient variation and thus a manifold capturing key axes of variation in different single-cell phenotypes among a large set of experimental conditions76.
遗传和化学扰动可显著影响细胞表型。现在可以进行大规模筛选,其中细胞受到不同遗传或化学扰动的干扰,然后在单细胞水平上进行分析。此类合并的扰动测序筛选 71 已用于通过基因敲除或激活筛选 72 来识别基因功能,并将编码和非编码变异分类为不同的扰动影响水平。此外,越来越大规模的 scRNA-seq 筛选可用来评估小分子药物 73 的作用。第一代分析通过测量基因 71 或细胞水平 74 的平均细胞反应来量化整体扰动效应。然而,不同扰动的结果可能在群体内异质细胞状态之间差异很大,例如无法通过建模平均值捕获的细胞行为。通过比较未受扰动和受扰动的细胞群体的分布,OT 可以捕获这种细粒度但异质的反应。为了捕捉扰动 k 对细胞群体影响的大小,我们可以计算未受扰动的细胞 X 和受扰动的细胞 Y k 的数据样本之间的 OT 距离 OT( X , Yk )。通过对已对齐的细胞状态进行比对,然后求和,OT 可以量化异质性细胞反应的强度(图 4a )。 基于这种直觉,Bunne 等人 6 通过 4i 多路复用 75 对两种黑色素瘤细胞系的 scRNA-seq 谱分析了不同抗癌药物的强度(图 4b、c )。检查不同抗癌药物治疗对不同细胞状态的 OT 成本以及所有细胞状态下的总 OT 成本,证明了 OT 距离如何可作为识别不同细胞状态对不同药物敏感性的度量。凋亡诱导剂(staurosporine)、蛋白酶体抑制剂(ixazomig 和 carfilzomib 或联合治疗 carfilzomib + pomalidomide + 地塞米松)、微管稳定剂(紫杉醇)和多种酪氨酸激酶的 ATP 竞争者,如 KIT 和 BCR-ABL(达沙替尼)在所有细胞状态下均引起显着的特征变化,因此显示出高运输成本(图 4c )。最后,OT 距离还可用于创建患者状态空间图,突出显示患者间变异的来源,从而形成一个流形,捕捉大量实验条件下不同单细胞表型变异的关键轴 76 。
图4:最佳运输距离在单细胞和空间生物学中的应用和结果。

a, The optimal transport (OT) distance can measure the strength of different perturbations, red and blue, by summing over the computed alignment between unperturbed μ and perturbed cells νblue and νred, respectively. b, For a mixture of two cell lines M130219 and M130429, the OT distance can be used to quantify the outcome of a single treatment, here for drugs trametinib and dabrafenib, for different subpopulations that are computed via Leiden clustering for individual features6, described through expression of markers pAKT and pERK. c, It can further be used to compare the strength of the response summarized over the entire population, here in a screen containing 35 different drugs6. d, The OT distance can also serve as a cell–cell similarity measure by computing it between the feature vectors of individual cells. e, The resulting pairwise distance matrix has a structure similar to metrics such as Pearson correlation22. f, Using OT as a cell–cell similarity metric results, however, in more coherent cell subpopulation clusters (as quantified through the silhouette score). g, Similarly, the OT distance can be used to model microenvironments (MEs) in spatial biology, in which each ME is represented by a collection of cells and their features. h,i, The MEs computed based on the OT distance not only comprise cells similar in the osmFISH uniform manifold approximation and projection space93 but also result in coherent clusters within the tissue (part i) that resemble the ground-truth tissue sectioning (part h)93. PCA, principal component analysis. Parts b and c adapted from ref. 6, Springer Nature Limited. Part e adapted with permission from ref. 22, Oxford University Press. Parts h and i adapted from ref. 93, Springer Nature Limited.
a 、最佳传输 (OT) 距离可以通过对未受干扰的 μ 和受干扰的细胞 ν 蓝色和 ν 红色之间计算出的比对求和来测量不同扰动(红色和蓝色)的强度。b 、 对于两种细胞系 M130219 和 M130429 的混合物,OT 距离可用于量化单次治疗的结果,这里针对药物曲美替尼和达拉非尼,针对不同的亚群,这些亚群是通过针对各个特征 6 的莱顿聚类计算得出的,通过标记 pAKT 和 pERK 的表达来描述。c、它还可以用于比较整个群体中总结的反应强度,这里是包含 35 种不同药物 6 的屏幕 。d 、OT 距离还可以通过在单个细胞的特征向量之间计算来用作细胞与细胞相似性度量。e 、 得到的成对距离矩阵具有类似于皮尔逊相关性 22 等指标的结构。 f 、然而,使用 OT 作为细胞与细胞相似性度量会产生更一致的细胞亚群簇(通过轮廓分数量化) 。g 、类似地,OT 距离可用于模拟空间生物学中的微环境(ME),其中每个 ME 由一组细胞及其特征表示。 h , i ,基于 OT 距离计算的 ME 不仅包含 osmFISH 均匀流形近似和投影空间 93 中相似的细胞,而且还会在组织内形成与真实组织切片(部分 h ) 93 相似的相干聚类(部分 i )。 PCA,主成分分析。部分 b 和 c 改编自参考文献 6 ,Springer Nature Limited。部分 e 经参考文献 22 许可改编,牛津大学出版社。部分 h 和 i 改编自参考文献 93 ,Springer Nature Limited。
OT can also serve as a distance metric between individual cells rather than cell populations and as such used to identify and distinguish between different cell states. Identifying specific cell types, states, programmes and contexts in which disease-implicated genes act is key to understanding biology both in homeostasis and in pathogenesis at the cell and tissue levels77 and a key motivation for the Human Cell Atlas initiative78. Classical strategies for identifying and characterizing cell heterogeneity typically often rely on unsupervised clustering79,80,81,82,83,84, whereby cells with similar features, such as gene expression profiles, are grouped based on a chosen notion of similarity and dimensionality reduction method. Owing to the curse of dimensionality, approaches use Euclidean or Manhattan distances on principal component analysis, uniform manifold approximation and projection85 or t-distributed stochastic neighbour embeddings86,87 or run Pearson correlation analysis on high-dimensional data88. Instead of relying on similarity metrics that ignore the structure or heterogeneity of a cell population, we can employ the OT distance as an alternative cell–cell similarity measure for cell-type identification. For this, we compute pairwise OT distances OT(xi, xj) (equation (4)) between the feature vectors xi and xj of each pair of cells i and j for cell state identification (Fig. 4d). The use of OT as a cell–cell similarity metric that, different from established approaches such as Pearson correlation (Fig. 4e), captures heterogeneous and continuous cell states across different data modalities has been demonstrated22. Extensions have further combined OT with deep metric learning to provide efficient cell–cell representations89,90.
OT 还可以作为单个细胞而不是细胞群之间的距离度量,因此用于识别和区分不同的细胞状态。识别与疾病相关的基因发挥作用的特定细胞类型、状态、程序和环境是理解细胞和组织水平上的体内平衡和发病机制生物学的关键 77 ,也是人类细胞图谱计划的主要动机 78 。识别和表征细胞异质性的经典策略通常依赖于无监督聚类 79 、 80 、 81 、 82 、 83 、 84 ,其中具有相似特征(例如基因表达谱)的细胞根据所选的相似性概念和降维方法进行分组。由于维数灾难,方法在主成分分析中使用欧几里得距离或曼哈顿距离、均匀流形近似和投影 85 或 t 分布随机邻域嵌入 86、87 , 或在高维数据 88 上运行皮尔逊相关分析。我们可以采用 OT 距离作为细胞类型识别的替代细胞间相似性度量,而不是依赖忽略细胞群体结构或异质性的相似性指标。为此,我们计算每对细胞 i 和 j 的特征向量 x 和 x 之间的成对 OT 距离 OT( x , x )(公式 ( 4 )),以进行细胞状态识别(图 4d )。 OT 作为细胞间相似性度量已被证明 22 ,与皮尔逊相关性(图 4e )等既定方法不同,它可以捕捉不同数据模态中异构且连续的细胞状态。扩展方法进一步将 OT 与深度度量学习相结合,以提供有效的细胞间表征 89 , 90 。
Spatial omics 空间组学
The development of new technologies for spatially resolved protein and RNA profiling has opened new opportunities for understanding the location-dependent properties of tissues, cells and molecules, as well as detecting cell–cell communication. A fundamental question in tissue biology is to recover the key structural/functional units of a tissue, in terms of multicellular communities, microenvironments (MEs) or ‘niches’. The OT distance can be used to analyse and characterize such MEs from spatially resolved data, enabling quantitative analysis of niches91,92,93,94,95,96,97,98. For this, we model the ME of each cell i by aggregating the feature vectors of its spatial neighbours into a histogram MEi. To understand distances or similarities to other cellular MEs, we compute the OT distance between all pairs of cellular MEs, in the form of OT(MEi, MEj) for all i, j (Fig. 4f). Subsequently, Yuan et al.93 and Mani et al.95 apply standard clustering approaches on the resulting pairwise distance matrix . Using multiplex fluorescence in situ RNA hybridization data, the detected MEs (Fig. 4g,h) resemble the ground-truth tissue section93 (Fig. 4i).
空间分辨蛋白质和 RNA 分析新技术的发展为理解组织、细胞和分子的位置依赖性以及检测细胞间通讯开辟了新的机会。组织生物学的一个基本问题是从多细胞群落、微环境 (ME) 或“生态位”的角度恢复组织的关键结构 / 功能单位。OT 距离可用于从空间分辨数据中分析和表征此类 ME,从而实现对生态位的定量分析 91、92 、 93 、94 、 95 、 96 、 97 、 98 。为此,我们通过将细胞 i 的空间邻居的特征向量聚合成直方图 ME 来建模每个细胞 i 的 ME。为了了解与其他细胞 ME 的距离或相似性,我们计算所有细胞 ME 对之间的 OT 距离,形式为所有 i 、 j 的 OT(ME, ME)(图 4f )。随后,Yuan 等人 93 和 Mani 等人 95 对得到的成对距离矩阵 运用标准聚类方法。使用多重荧光原位 RNA 杂交数据,检测到的 ME(图 4g,h )与真实组织切片 93 (图 4i )相似。
Alignment in single-cell and spatial omics
单细胞和空间组学的比对
OT has an even more prominent role in both single-cell and spatial biology as an approach to align between point clouds via the OT plan P.
OT 作为一种通过 OT 计划 P 在点云之间进行对齐的方法,在单细胞和空间生物学中发挥着更为突出的作用。
Single-cell omics 单细胞组学
The first and still most eminent result of OT in single-cell biology employs the OT plan to reconstruct the temporal trajectories of cells over the course of differentiation, a setting similar to Example 1. Cellular differentiation is accompanied by both molecular and morphological changes, which both drive the process and respond to it. Molecular characterization of the differentiation processes and understanding the extrinsic and intrinsic guiding programmes of cells remain fundamental challenges in developmental biology. Because the process involves inherent diversification of a cell population and is not perfectly synchronous, approaches relying on the bulk profiling of cell populations fall short in tackling two key obstacles: identifying various cell types within a population and tracking the development of each of these types. Single-cell omics methods partially address these challenges by profiling individual cells, but their destructive nature impedes the direct tracking of cell fates from ancestors to their descendants (Example 1).
OT 在单细胞生物学中的第一个也是最突出的成果是采用 OT 方案重建细胞在分化过程中的时间轨迹,类似于示例 1 的设置。细胞分化伴随着分子和形态的变化,这些变化既驱动该过程,也对其作出反应。分化过程的分子表征以及理解细胞的内在和外在指导程序仍然是发育生物学的基本挑战。由于该过程涉及细胞群体固有的多样化并且并非完全同步,因此依赖于对细胞群体进行批量分析的方法无法解决两个关键障碍:识别群体中的各种细胞类型并跟踪每种类型的发育。单细胞组学方法通过分析单个细胞部分解决了这些挑战,但它们的破坏性阻碍了从祖先到后代的细胞命运的直接追踪(示例 1)。
Previous tools aiming to reconstruct cellular dynamics from time-resolved snapshot data often rely on strong constraints imposed by nearest neighbour graphs99,100,101,102, restrict themselves to modelling population averages over time103 or fall short in considering cellular growth and death in developmental processes104. Instead, OT is uniquely suited for the challenge of modelling the continuous emergence of different cell types and branching events by reconstructing a fate map from time-resolved single-cell measurements. Given a cell with a specific profile at a time point, OT enables determination of which descendants it is likely to have at a later time point and which ancestors it had at an earlier time point (Fig. 5a). Approaching this problem with an OT framework was first studied in the context of reconstructing differentiation during reprogramming of fibroblasts to induced pluripotent stem cells5, from >315,000 mouse embryonic fibroblasts (MEFs) profiled along 18 time points (Fig. 5b). Cells at time point t are connected to their ancestors at time t − 1, by finding the corresponding transport plan Pt−1,t between each pair of consecutive time steps (Fig. 5a). Using entropy regularization when computing the transport plan further provides a notion of statistical uncertainty in the inferred descendant distribution (Box 4). Employing an unbalanced OT problem further accounts for cell division and death5,62,64,105. OT plans then allow tracing stem and progenitor cell differentiation through a series of fate decisions, marked by a continuous adaptation of cells that refine their identity until reaching a functional end state. This allows the tracking of changes in gene expression trends along different lineages, such as stromal and mesenchymal-to-epithelial transition cell states (Fig. 5c), or to relate likely to cellular ancestors and descendants over time (Fig. 5d). Finally, we can compress the sequence of transport plans into a single fate transition table that indicates into which lineages various cell fates are developing (Fig. 5e).
图 5:最优运输计划在单细胞和空间生物学中的应用和结果。

a, The optimal transport (OT) plan P re-aligns cells from consecutive measurements μt and μt+1 at time points t and t + 1 based on the feature vector of each cell. This allows for tracing the developmental origin of different cells throughout their differentiation process. b, An induced pluripotent stem (iPS) cell reprogramming experiment conducted with separate snapshot measurements across 18 days gives rise to a diverse set of cell types5. c, The OT plan enables tracing gene signature trends along trajectories to stromal and mesenchymal-to-epithelial transition (MET), here captured through mouse embryonic fibroblast (MEF) identity, secretory phenotype (SASP), proliferation and epithelial signatures5. d, It further allows us to trace the developmental history of each cell, as exemplified by visualizing the ancestors of day 18 stromal cells in serum in a force-directed layout embedding. Colour denotes the day and intensity denotes the probability. e, The OT plan can be further compressed into a fate transition table that indicates differentiation from and to different cell types5. f, Besides, the OT plan allows for spatially reconstructing tissues. To recover the tissue structure from gene expression measurements, one can employ the Gromov–Wasserstein plan to map each expression vector onto a reference atlas provided. g,h, Taking the spatial gene expression of the genes SNA, KEN and EVE of the Drosophila embryo as an example (part g) and provided with a reference atlas, here of the Drosophila embryo19, the OT plan can be used to spatially reconstruct gene expression patterns along the reference atlas19 (part h). Parts b–d adapted with permission from ref. 5, Elsevier. Parts f and h adapted from ref. 19, Springer Nature Limited.
a 、最优传输 (OT) 计划 P 根据每个细胞的特征向量,对时间点 t 和 t + 1 处连续测量 μ t 和 μ t +1 中的细胞进行重新排列。这可以追踪不同细胞在整个分化过程中的发育起源。b 、 在 18 天内使用单独快照测量进行的诱导多能干细胞 ( iPS ) 重编程实验产生了多种细胞类型 5。c 、OT 计划可以追踪沿着基质和间质-上皮转化 (MET) 轨迹的基因特征趋势,这里通过小鼠胚胎成纤维细胞 (MEF) 身份、分泌表型 (SASP)、增殖和上皮特征 5 捕获 。d 、它进一步使我们能够追踪每个细胞的发育历史,例如通过在力导向布局嵌入中可视化血清中第 18 天基质细胞的祖先。颜色表示日期,强度表示概率。e 、 OT 计划可以进一步压缩为一个命运转换表,用于指示与不同细胞类型的分化 5。f 、 此外,OT 计划还允许空间重建组织。为了从基因表达测量中恢复组织结构,可以采用 Gromov-Wasserstein 计划将每个表达载体映射到提供的参考图谱上。 g , h ,以果蝇胚胎的 SNA 、 KEN 和 EVE 基因的空间基因表达为例(部分 g ),并提供参考图谱(此处为果蝇胚胎 19 ),OT 计划可用于沿着参考图谱 19 在空间上重建基因表达模式(部分 h )。部分 b - d 经 Elsevier 许可改编自参考文献 5 。部分 f 和 h 改编自 Springer Nature Limited 的参考文献 19 。
The inferred trajectories can be validated in various ways. Following the scientific method, the inferred trajectories can be used to generate hypotheses, which are then tested experimentally. For example, in reprogramming OT trajectories identified cell signalling factors that could increase the efficiency of reprogramming, which were then tested in laboratory experiments to assess whether the predicted effect was seen5. Another direct, data-driven approach to validate OT trajectories is through geodesic flow. Given three time points, one can connect time point 1 directly to time point 3 (holding out data from time point 2) and verify how close interpolated trajectories are to the held-out data5,106. When additional measurements can be made, OT trajectories can be compared to assess consistency. For example, barcoded data sets107, transcriptome profiling approaches that preserve cell viability such as Live-Seq108, live microscopy109 or Raman microscopy110 can be used to evaluate the corresponding transport plan.
推断的轨迹可以通过多种方式进行验证。按照科学方法,推断的轨迹可用于生成假设,然后通过实验进行测试。例如,在重新编程 OT 轨迹中确定了可以提高重新编程效率的细胞信号传导因子,然后在实验室实验中对这些因子进行测试,以评估是否看到了预测的效果 5 。另一种直接的、数据驱动的验证 OT 轨迹的方法是通过测地线流 。给定三个时间点,可以将时间点 1 直接连接到时间点 3(保留时间点 2 的数据)并验证内插轨迹与保留数据的接近程度 5,106 。 当可以进行其他测量时,可以比较 OT 轨迹以评估一致性。例如,可以使用条形码数据集 107 、保留细胞活力的转录组分析方法(如 Live-Seq 108 ) 、活体显微镜 109 或拉曼显微镜 110 来评估相应的运输计划。
Example 3: spatial reconstruction of cell populations
示例3:细胞群的空间重建
The OT plan also allows mapping of biological processes in space, as illustrated by a third example. Most high-throughput single-cell profiling methods require tissue dissociation, such that the location zi of cell i with molecular profile xi in the original tissue is unknown. In some cases, however, previous knowledge of the tissue structure, or a limited amount of spatially resolved data, for a set of spatial landmarks or through spatial transcriptomics may be provided, giving access to either the location of the reference atlas , with zi denoting the location of a grid cell i on a reference atlas of marker genes (Fig. 1c), or a limited number of spatially resolved cells of similar origin , where for cell i both a transcriptomic profile and tissue location are known. To spatially reconstruct tissues or assign cells onto a spatial location in a reference atlas, an alignment P between X and is required.
OT 计划还允许在空间中映射生物过程,如第三个示例所示。大多数高通量单细胞分析方法需要组织分离,因此原始组织中具有分子谱的细胞位置未知。然而,在某些情况下,可以提供先前的组织结构知识,或一组空间标志或通过空间转录组学获得的有限量的空间解析数据,从而可以访问参考图谱的位置。 ,表示标记基因参考图谱上网格细胞的位置(图 ),或有限数量的相似起源的空间分辨细胞 ,其中细胞的转录组谱 和组织位置 是已知的。为了在空间上重建组织或将细胞分配到参考图谱中的空间位置,需要对 和 进行对齐 是必需的。
Spatial omics 空间组学
Because Example 3 is a problem between heterogeneous spaces — non-spatially resolved data X and a reference atlas or a limited amount of spatially resolved data — classic OT methods do not work. Instead, we can use the transport plan resulting from the Gromov–Wasserstein objective (equation (12)). This characteristic was first used to create a cartography gene expression20: an OT-based framework for de novo spatial reconstruction of single-cell gene expression with little or no previous knowledge. At the core of this framework lies a structural correspondence hypothesis that cells in physical proximity share similar gene expression profiles. Taking the pairwise distances of both gene expression (c(xi, xj) between cells i and j) and target atlas space ( between locations k and l between reference atlas locations), the OT plan then maps a single-cell xi to a particular location in the reference atlas yk (ref. 19) (Fig. 5f). For example, using spatial expression data in the Drosophila embryo (Fig. 5g) and a Drosophila embryo reference atlas (Fig. 5f), this approach reconstructs spatial expression patterns from non-spatially resolved scRNA-seq data using the Gromov–Wasserstein plan P (equation (12)) (Fig. 5h).
因为示例 3 是异质空间之间的问题——非空间解析数据和参考图集或有限数量的空间解析数据 — 经典的 OT 方法不起作用。相反,我们可以使用由 Gromov–Wasserstein 目标函数(方程())得出的传输规划。这一特性最初用于创建基因表达制图:一个基于 OT 的框架,用于在几乎或完全没有先前知识的情况下从头空间重建单细胞基因表达。该框架的核心是一个结构对应假说,即物理上接近的细胞具有相似的基因表达谱。取细胞和之间的基因表达((,))和目标图谱空间( 位置之间和参考图谱位置之间),OT 计划随后将单细胞映射到参考图谱中的特定位置(参考文献)(图)。例如,使用胚胎中的空间表达数据(图)和胚胎参考图谱(图),该方法使用 Gromov–Wasserstein 计划(方程 ())(图)从非空间解析的 scRNA-seq 数据重建空间表达模式。
When combining the classic OT objective (equation (3)) and the extension to heterogeneous spaces (equation (12)), the resulting problem is known as fused Gromov–Wasserstein objective111. The coupling P resulting from the fused Gromov–Wasserstein objective can be used to pairwise align slices of spatial transcriptomics measurements, such as (ref. 112). In this setting, P optimizes the alignment based on both expression feature similarity between X and and physical distance between spots on a spatial transcriptomics slide Z and . Spot i on one slice is mapped to spot j with weight Pij if expression profile xi is similar to expression profile xj, and if a pair of spots i, k in one slide that is mapped to a pair of spots j, l on the other slide with weights Pij and Pkl, then the spatial distance c(yi, yk) is close to . The resulting pairwise aligned spatial transcriptomics slides can be combined into a stacked 3D alignment of a tissue as well as integrated into a single tissue slice112.
当将经典 OT 目标(方程 ( 3 ))与扩展到异质空间(方程 ( 12 ))相结合时,所产生的问题称为融合 Gromov-Wasserstein 目标 111 。由融合 Gromov-Wasserstein 目标产生的耦合 P 可用于成对比对空间转录组学测量的切片,例如 (参考文献 112 )。在这种情况下, P 根据 X 和 之间的表达特征相似性以及空间转录组学载玻片 Z 和 上点之间的物理距离来优化比对。如果表达谱 x 与表达谱 x 相似,则将一个切片上的点 i 映射到具有权重 P i j 的点 j ;如果一张载玻片上的一对点 i , k 映射到另一张载玻片上的一对点 j , l ,权重分别为 P i j 和 P k l ,则空间距离 c ( y , y k ) 接近于 。所得的成对比对的空间转录组学载玻片可以组合成组织的堆叠 3D 比对,也可以集成到单个组织切片 112 中。
OT map between different cell populations
不同细胞群之间的 OT 图
Beyond the OT plan, OT also provides a map T that maps between distributions, for example, different cell populations μ and ν. With the growing availability of large-scale data sets8,113,114,115, recent efforts have concentrated on inferring T from data. Crucially, parameterizing T — either through the Sinkhorn algorithm (equation (6)) (Box 4) or via neural networks — allows us to map a point x from source distribution μ to y = T(x) of target distribution ν out-of-sample. This generalization to unseen data points enables prediction of the transition for a new unobserved sample xnew, without the need to recompute map T for the new instances. This is particularly important for applications in precision medicine: given a previously unobserved patient, it is possible to forecast how their cells, obtained through biopsies or from tissue culture, might respond to a subsequent therapy, by using a map T learned on a previous patient cohort6. Such predictions could be made for different potential treatments to select the most effective one for the patient. Because cell populations and their response to treatments are often heterogeneous, with different cell types and states exhibiting distinct responses to a drug, tackling the problem on the level of distributions as done in OT presents a particular expressive modelling approach.
除了 OT 计划之外,OT 还提供了一个映射分布之间的图 T ,例如,不同的细胞群 μ 和 ν 。随着大规模数据集 8、113、114、115 的日益普及,最近的努力集中在从数据中推断 T。 至关重要的是,参数化 T — — 无论是通过 Sinkhorn 算法(公式 ( 6 ))(框 4 )还是通过神经网络 — — 都使我们能够将点 x 从源分布 μ 映射到目标分布 ν 的 y = T ( x )。这种对看不见的数据点的推广使得能够预测新的未观察样本 x new 的转变,而无需为新实例重新计算图 T。 这对于精准医疗中的应用尤为重要:给定一个以前未观察的患者,可以通过使用在先前患者队列 6 上学习到的图 T 来预测通过活检或组织培养获得的细胞对后续治疗的反应。此类预测可用于预测不同的潜在治疗方法,从而为患者选择最有效的方案。由于细胞群体及其对治疗的反应通常具有异质性,不同类型的细胞和状态对药物的反应也各不相同,因此,像在 OT 中那样在分布层面上解决问题,可以提供一种富有表现力的建模方法。
Similarly, this has fruitful applications for predicting the effect of unseen (combinations of) genetic perturbations, in particular with the rise of Perturb-seq screens that allow to simultaneously profile the effect of a perturbation on individual cells71,72,74. By leveraging the learned transport map conditioned on the desired genetic perturbation, researchers can anticipate the consequences of genetic alterations on cellular states27,116. This not only enhances our understanding of cellular behaviour under different genetic conditions but also allows in silico predictions for regimes where the number of (combinations of) perturbations exceeds experimental capacity.
类似地,这对于预测看不见的(组合)遗传扰动的影响也有着卓有成效的应用,特别是随着 Perturb-seq 筛选的兴起,这种筛选可以同时分析扰动对单个细胞的影响 71 、 72 、 74 。通过利用以所需遗传扰动为条件的学习到的传输图,研究人员可以预测遗传改变对细胞状态的影响 27 、 116 。这不仅增强了我们对不同遗传条件下细胞行为的理解,而且还允许对扰动数量(组合)超过实验能力的状态进行计算机预测。
Given a data set with unaligned samples X of the untreated population μ and samples Y of the treated population ν (Example 1), we aim to learn a map Tk that predicts the treated state y = Tk(xi) of each cell xi, given a drug k. Several strategies have been proposed for parameterizing and learning T (refs. 6,7,50,56,57,117,118). These include approaches that parameterize the dual potentials (6) via neural networks, such that T = ∇fθ (refs. 6,56,57,118) (Fig. 6a), directly Tθ given either an additional regularizer that quantifies whether Tθ agrees with theoretical properties of OT7 or via a scaling factor to model the unbalanced OT problem17.
给定一个数据集 , 其中包含未治疗群体 μ 的未对齐样本 X 和已治疗群体 ν 的样本 Y (例 1),我们的目标是学习一个映射 T k ,该映射预测给定药物 k 时每个细胞 x 的治疗状态 y = T k ( x )。已经提出了几种参数化和学习 T 的策略(参考文献 6、7、50、56、57、117、118 ) 。 这些包括通过神经网络参数化对偶势 ( 6 ) 的方法,使得 T = ∇ f θ ( 参考文献 6、56、57、118 ) (图 6a ),直接给定 T θ ,要么给出一个额外的正则化器来量化 T θ 是否符合 OT 的理论性质 7 ,要么通过一个缩放因子来模拟不平衡的 OT 问题 17 。
图6:最佳传输图在单细胞生物学中的应用和结果。

a, The optimal transport (OT) map enables us to predict the outcome and effect of different perturbations for single cells. By learning these maps using a neural network, we are able to infer the perturbed cell states (red and blue) even for new and unseen cells, such as those from novel cell types or patients. b,c, Contrary to the average map6 (part b), such neural OT maps capture fine-grained perturbation responses of heterogeneous single-cell populations (part c). The distribution of control cells is denoted by blue, treated cells by grey and the resulting map is indicated through arrows6. PCA, principal component analysis. Parts b and c adapted from ref. 6, Springer Nature Limited.
a 、最佳传输(OT)图使我们能够预测不同扰动对单个细胞的结果和影响。通过使用神经网络学习这些图,我们能够推断出扰动的细胞状态(红色和蓝色),即使是新的和看不见的细胞,例如来自新细胞类型或患者的细胞。b 、 c 、与平均图 6 (部分 b )相反,这种神经 OT 图捕获了异质单细胞群的细粒度扰动响应(部分 c )。对照细胞的分布用蓝色表示,处理过的细胞用灰色表示,得到的图用箭头 6 表示。PCA,主成分分析。b 部分和 c 部分改编自参考文献 6 ,Springer Nature Limited。
For example, given a mixture of two melanoma cell lines, neural OT solvers reconstructed heterogeneous responses to different cancer drugs6. The learned map (Fig. 6a,c), or vector field, explains for each cell state, for example, the location in the projected data space, how unperturbed cells transform into cells perturbed by a treatment. Conversely, a map capturing only the average treatment effect would apply the same effect to each cell state, independent of the location in the data space or the feature representation of each cell, and is thus less well suited to model the true diversity of the biological phenomenon (Fig. 6b). Unlike previous approaches14,119,120,121, neural OT schemes encode important inductive biases that facilitate learning and result in a reliable and easy-to-train framework and demonstrate consistently strong performance on a wide span of applications6,7.
例如,给定两种黑色素瘤细胞系的混合物,神经 OT 求解器重建了对不同抗癌药物的异质响应 6 。学习到的图(图 6a、c )或矢量场解释了每个细胞状态,例如,在投影数据空间中的位置,未受干扰的细胞如何转变为受治疗干扰的细胞。相反,仅捕获平均治疗效果的图将对每个细胞状态应用相同的效果,而与数据空间中的位置或每个细胞的特征表示无关,因此不太适合模拟生物现象的真实多样性(图 6b )。与以前的方法 14、119、120、121 不同 , 神经 OT 方案编码了重要的归纳偏差,这些偏差有助于学习并产生可靠且易于训练的框架,并在广泛的应用中表现出始终如一的强劲性能 6、7 。
Applications 应用
OT is poised to become a workhorse of modern biological analysis owing to its versatility in studying systems in space and time, as well as the availability of efficient numerical algorithms and deep learning frameworks. To illustrate this, we highlight key studies using the OT framework to address problems in cell differentiation, prediction of response to perturbations, multimodal integration and spatial reconstruction.
OT 凭借其在研究空间和时间系统方面的多功能性,以及高效的数值算法和深度学习框架,有望成为现代生物分析的主力军。为了说明这一点,我们重点介绍了一些使用 OT 框架解决细胞分化、扰动响应预测、多模态积分和空间重建等问题的关键研究。
Reconstructing cell differentiation processes
重建细胞分化过程
Drawing from the metaphor of Waddington’s landscape122, developmental biology commonly describes cell differentiation as marbles rolling down a complex landscape122. Each valley within this landscape represents a specific differentiated fate that a cell might take, with the depth of the valley signifying the stability of the state123. Paths on the developmental manifold then describe the evolution of a time-varying probability distribution on a high-dimensional expression space, representing the continuous changes in cell profiles over time. We have reviewed earlier the first method to approximate such dynamic processes through a sequence of OT plans that are computed between distinct snapshots5. As the field has developed, later approaches have taken advantage of the flexibility of OT and extended the models to heterogeneous spaces, dynamic formulations and continuous cellular dynamics. This is achieved by incorporating differential equations, additional experimental data and deep learning methods.
借用沃丁顿景观 122 的比喻,发育生物学通常将细胞分化描述为弹珠在复杂的景观 122 上滚落。景观中的每个山谷代表细胞可能采取的特定分化命运,山谷的深度表示状态的稳定性 123 。发育流形上的路径描述了高维表达空间上随时间变化的概率分布的演变,表示细胞概况随时间的连续变化。我们之前已经回顾了第一种方法,通过在不同快照 5 之间计算的一系列 OT 计划来近似这种动态过程。随着该领域的发展,后来的方法利用 OT 的灵活性,并将模型扩展到异构空间、动态公式和连续细胞动力学。这是通过结合微分方程、额外的实验数据和深度学习方法实现的。
Although cellular dynamics have been reconstructed purely based on gene expression information5,123, the framework introduced in Example 1 has been expanded by integrating both lineage and expression information across temporally resolved snapshots, enabled by new laboratory-based methods to track cell lineages10,124,125. First employed in Caenorhabditis elegans10, Lange et al.124 use the fused Gromov–Wasserstein distance (equation (12)) to not only align cells from consecutive time points based on minimizing differences in their gene expression features but also with respect to maintaining consistency across intra-individual lineage relations and inter-individual gene expression. Such an analysis, however, does not generalize to lineage trees that are not completely deterministic, as in, where the lineage tree used at a consecutive time point is not a perfect extension of the lineage tree in the previous time point, and is thus restricted to simple organisms such as C. elegans but not mammals.
尽管细胞动力学已完全基于基因表达信息 5、123 重建,但示例 1 中引入的框架已通过整合时间分辨快照中的谱系和表达信息而得到扩展,并通过新的基于实验室的方法来追踪细胞谱系 10、124、125。Lange 等人 124 首次将该方法用于秀丽隐杆线虫 10 ,他们使用融合的 Gromov-Wasserstein 距离(方程 ( 12 )),不仅可以根据最小化基因表达特征的差异来对来自连续时间点的细胞进行对齐,而且还可以保持个体内谱系关系和个体间基因表达的一致性。然而,这种分析并不适用于不完全确定性的谱系树,例如,在连续时间点使用的谱系树不是前一时间点谱系树的完美延伸,因此仅限于简单生物,如秀丽隐杆线虫 ,但不适用于哺乳动物。
The proposed approaches highlighted so far have only provided a coarse approximation of continuous developmental processes, through pairwise alignments between consecutive snapshots5. To overcome this limitation, recent efforts have concentrated on reconstructing continuous dynamics from snapshot measurements by building on the dynamic formulation of OT and its connections to PDE and SDE43. One approach13 establishes a link between the Benamou–Brenier43 formulation (equation (15)) and continuous normalizing flows, also known as neural ordinary differential equations55,126, to model paths of cell differentiation over time. This approach was used, for example, to model and interpolate between scRNA-seq measurement time points in a system of differentiating embryoid bodies127.
到目前为止,所强调的建议方法仅通过连续快照 5 之间的成对比对提供了对连续发育过程的粗略近似。为了克服这一限制,最近的努力集中在通过构建 OT 的动态公式及其与 PDE 和 SDE 43 的联系,从快照测量中重建连续动力学。一种方法 13 在 Benamou-Brenier 43 公式(方程( 15 ))和连续归一化流(也称为神经常微分方程 55,126 ) 之间建立联系,以模拟细胞随时间的分化路径。例如,这种方法用于在分化胚状体的系统中对 scRNA-seq 测量时间点进行建模和插值 127 。
Because both cell fate decisions and the underlying measurements are inherently stochastic, a large body of work now employs SDEs as the model backbone. Concretely, the entropy-regulated OT problem (equation (19))25 coincides with the famous Schrödinger bridges concept128,129, which optimizes for the stochastic process that best describes the evolution of a population μ0 at time point 0 to a population μT at time point T, given some reference process or previous knowledge on the underlying dynamical system (for example, Brownian motion). The solution of the Schrödinger bridge is a system of SDEs, and recent numerical12,16 and neural-network-based69,130,131 approaches have been proposed for utilizing Schrödinger bridges to reconstruct cellular differentiation processes over time11,109,132,133,134 with extensions to allow for birth and death events62. An added benefit is that this viewpoint leads to rigorous theoretical guarantees for trajectory inference12, which otherwise mostly lack for differentiation reconstruction methods.
由于细胞命运决策和底层测量本质上都是随机的,因此目前大量研究采用随机微分方程 (SDE) 作为模型支柱。具体而言,熵调节的 OT 问题(方程 ( 19 )) 25 与著名的薛定谔桥概念 128 , 129 相吻合,该概念针对的是随机过程进行优化,该过程最能描述种群 μ 0 在时间点 0 到时间点 T 的演化 ,给定一些参考过程或关于底层动力系统的先验知识(例如,布朗运动)。薛定谔桥的解决方案是一组 SDE,最近提出了数值 12、16 和基于神经网络 69、130、131 的方法 ,利用薛定谔桥重建细胞随时间的分化过程 11、109、132、133、134 , 并扩展至考虑出生和死亡事件 62。 另一个好处是,这种观点为轨迹推断 12 提供了严格的理论保证,而这在分化重建方法中大多是缺乏的。
Instead of reconstructing the underlying stochastic process, some methods approach modelling cell differentiation through estimating an underlying energy that guides the overall dynamics9,15,135,136. This can be achieved by taking advantage of the connection of OT to gradient flows137 and PDEs138 building up on the Jordan, Kinderlehrer and Otto scheme139 (also known as JKO flows)140,141,142,143.
一些方法不是重建潜在的随机过程,而是通过估计引导整体动力学的潜在能量来模拟细胞分化 9 、 15 、 135 、 136 。这可以通过利用 OT 与梯度流 137 和基于 Jordan、Kinderlehrer 和 Otto 方案 139 (也称为 JKO 流) 140 、 141 、 142 、 143 建立的 PDE 138 的连接来实现。
OT can further be used as a loss function to assess how well the chosen dynamic model approximates the experimental measurements. Concretely, Hashimoto et al.144 assume a Langevin dynamic for the evolving cells, driven by the gradient flow of a (neural) energy function, where the parameters of that energy are estimated through regularized OT distances between the predictions of the model and the corresponding ground-truth snapshots25. Furthermore, approaches connecting autoencoders14,145 or generative adversarial models17,125 with OT have similarly successfully reconstructed cell differentiation processes from snapshot measurements.
OT 可以进一步用作损失函数,以评估所选动态模型与实验测量值的近似程度。具体而言,Hashimoto 等人 144 假设进化细胞呈现朗之万动力学,由(神经)能量函数的梯度流驱动,其中该能量的参数通过模型预测值与相应真实快照 25 之间的正则化 OT 距离来估计。此外,将自编码器 14、145 或生成对抗模型 17、125 与 OT 连接起来的方法同样能够从快照测量值中成功地重建细胞分化过程。
Predicting single-cell responses
预测单细胞反应
Several methods predicting the responses to perturbations with genetics or small-molecule drugs employ OT in their core. When the effect of the perturbation on the molecule profile is monitored at one or more (reasonably proximal) time points following the perturbation, OT allows us to reconstruct the incremental changes in the molecular profile of each cell introduced through perturbations. One important biological question that has been tackled by OT in this context is to predict the outcome of perturbations out-of-sample in new biological contexts, such as a new cell type or individual. To tackle this challenge, neural OT solvers are trained from unaligned, unperturbed and perturbed samples, such as cells measured before and after drug treatment or genetic perturbation. Once the parameterized OT map is optimized, these methods infer the perturbation effect on unseen cells6,7. Further work accounts for cell growth and death by extending these frameworks to partial matchings18,62,105. Moreover, as patients might respond differently to a particular treatment depending on factors such as treatment history, genetic status or other meta-information, several studies developed a neural OT scheme that can be conditioned on such contexts27. Such generalizations then provide a neural OT framework for modelling fine-grained responses that can be conditioned on factors such as the applied perturbation dosage or the specific perturbation of interest. Tackling the problem on the distributional level enables us to model heterogeneous cell responses6,7. This represents a strong modelling advantage over previous methods that model perturbation responses through a single arithmetic operation, either in a learned low-dimensional embedding119,120,121,146 or parameterized through graph neural networks116.
有几种方法利用遗传学或小分子药物预测对扰动的反应,其核心是 OT。当在扰动后的一个或多个(合理接近的)时间点监测扰动对分子谱的影响时,OT 使我们能够重建通过扰动引入的每个细胞的分子谱的增量变化。在此背景下,OT 已经解决的一个重要生物学问题是预测新生物背景下样本外扰动的结果,例如新的细胞类型或个体。为了应对这一挑战,神经 OT 求解器从未对齐、未扰动和扰动样本(例如在药物治疗或遗传扰动前后测量的细胞)进行训练。一旦参数化的 OT 图被优化,这些方法就可以推断出扰动对看不见的细胞的影响 6、7 。 进一步的研究通过将这些框架扩展到部分匹配 18、62、105 来解释细胞的生长和死亡。此外,由于患者对特定治疗的反应可能因治疗史、遗传状况或其他元信息等因素而异,一些研究开发了一种可根据此类背景进行条件调节的神经 OT 方案 27。 这种概括随后提供了一个神经 OT 框架,用于建模细粒度响应,这些响应可根据所施加的扰动剂量或感兴趣的特定扰动等因素进行条件调节。在分布层面上解决这个问题使我们能够对异质细胞反应进行建模 6、7 。 这代表了比以前通过单一算术运算对扰动响应进行建模的方法更强的建模优势,无论是在学习到的低维嵌入 119、120、121、146 中 , 还是通过图神经网络 116 进行参数化。
Causal inference — the process of determining the cause-and-effect relationships between variables or events based on observed data — has been connected to OT. In particular, in classical treatment and control study design, determining causal relationships between two variables is a fundamental and challenging task, allowing us to answer questions around patient outcomes after receiving different treatments and gene knockdown147,148. Contrary to previous (often linear) approaches designed to estimate average (or aggregate) causal effects149, the concept of using OT to measure the discrepancy between observed and counterfactual distributions has been proposed, providing a quantitative measure of causality147. When combined with active learning approaches, these frameworks allow the nomination and validation of high-confidence causal hypotheses in therapy design and treatment planning. This allows, for example, the identification and prioritization of transient but causally active candidate drug targets from single-cell observations, guiding efficient and cost-effective in vivo validations150.
因果推断——根据观察到的数据确定变量或事件之间因果关系的过程——与 OT 相关。尤其是在经典的治疗和对照研究设计中,确定两个变量之间的因果关系是一项基本且具有挑战性的任务,它使我们能够回答患者在接受不同治疗和基因敲减后的结果问题 147、148 。 与以前用于估计平均(或总体)因果效应 149 的方法(通常是线性的)相反,人们提出了使用 OT 来衡量观察到的分布和反事实分布之间差异的概念,从而提供了因果关系的定量测量 147。 当与主动学习方法相结合时,这些框架允许在治疗设计和治疗计划中提名和验证高置信度的因果假设。例如,这可以从单细胞观察中识别和确定短暂但有因果活性的候选药物靶点的优先级,从而指导高效且经济的体内验证 150 。
Multimodal omics integration
多模态组学整合
Ongoing advances in single-cell spatial genomics have moved the field rapidly from measurements of a single modality (RNA in cells and often protein in tissues) to multimodal measurements across many molecular (DNA, chromatin, RNA, proteins and their modifications) and morphological/histological levels, in either the same cell and tissue simultaneously, or in related but separate samples. Although each modality provides a different perspective on cell states and underlying mechanisms, their integration is critical for studying cell and tissue identity and function.
单细胞空间基因组学的持续进展已使该领域从单一模态(细胞中的 RNA,通常指组织中的蛋白质)的测量迅速发展到涵盖多个分子(DNA、染色质、RNA、蛋白质及其修饰)和形态/组织学水平的多模态测量,这些测量既可在同一细胞和组织中同时进行,也可在相关但独立的样本中进行。尽管每种模态都为理解细胞状态和潜在机制提供了不同的视角,但它们的整合对于研究细胞和组织的特性和功能至关重要。
When each modality is measured separately, obtaining a unified multimodal model of the same biological system requires aligning measurements across multiple modalities (Example 2 and Fig. 1d). Each technology, however, is adjusted through distinct parameters, subject to different sources of noise, and records features that may be nominally incomparable with those from other technologies, making a straightforward integration challenging. Many approaches now utilize autoencoder to learn a common representation across distinct data modalities, making it possible to compare and integrate data from different sources151,152,153. Similarly, as described earlier, OT can be used for harmonizing and pairing multimodal data. Using the Gromov–Wasserstein approach, the alignment is computed based on matching intramodality distances and aims to preserve the local geometry of each single-cell data set21. Recent advances unified the two approaches, integrating a coupled variational autoencoder and partial OT, such that the resulting latent space aligns the single-cell distributions of different modalities154. This cross-modality alignment was extended between profiling technologies such as sequencing and high content cell imaging155. By expanding the Gromov–Wasserstein formulation by a constraint on the cross-modality cellular coupling matrix, the method further allows integration and accounting for label information (such as the corresponding perturbation or a cell-type annotation)155. Similar extensions to partial matchings156,157 are able to handle disproportionate cell-type representation and differing sample sizes across single-cell measurements.
当单独测量每种模态时,要获得同一生物系统的统一多模态模型,需要跨多种模态对齐测量值(示例 2 和图 1d )。然而,每种技术都通过不同的参数进行调整,受到不同噪声源的影响,并记录可能与其他技术名义上无法比较的特征,这使得直接集成具有挑战性。现在许多方法利用自动编码器来学习不同数据模态之间的共同表示,从而可以比较和整合来自不同来源的数据 151、152、153 。类似地,如前所述,OT 可用于协调和配对多模态数据。使用 Gromov-Wasserstein 方法,对齐是基于匹配模态内距离计算的,旨在保留每个单细胞数据集的局部几何形状 21 。最近的进展统一了这两种方法,集成了一个耦合变分自动编码器和部分 OT,使得得到的潜在空间可以对齐不同模态的单细胞分布 154 。这种跨模态比对在测序和高内涵细胞成像等分析技术之间得到了扩展 155 。通过对跨模态细胞耦合矩阵进行约束来扩展 Gromov–Wasserstein 公式,该方法进一步允许整合和解释标签信息(例如相应的扰动或细胞类型注释) 155 。 与部分匹配 156、157 类似的扩展能够处理单细胞测量中不成比例的细胞类型表示和不同的样本大小。
Other technological advances allow the simultaneous profiling of multiple modalities from the same cell, such as the measurement of chromatin accessibility and gene expression158. Although in such cases no re-aligning of distinct cells across different modalities is required, these modalities profile distinct cellular processes that take place across multiple layers and times. For example, changes in histone modifications and transcription factor binding may precede changes in gene expression, creating chromatin states that bias genes for activation or repression to alter lineage outcomes158,159,160. To detect such phenomena, the development of multiview learning methods is crucial. To fill the gap, OT allows for the integration of paired multi-omics data161 and cross-modality inference23,24. Finally, OT can also be used for atlas creation, batch correction47,162 or multiscale integration of data sets containing a single or multiple data modalities (for example, single-cell genomics and digital pathology163) of patient cohorts164,165,166,167. This uncovers structure and heterogeneity as well as similarities between multiscale representations of patients168.
其他技术进步允许同时分析来自同一细胞的多种模态,例如测量染色质的可及性和基因表达 158 。尽管在这种情况下不需要跨不同模态重新排列不同的细胞,但这些模态分析了跨多层和多时间发生的不同细胞过程。例如,组蛋白修饰和转录因子结合的变化可能先于基因表达的变化,从而产生染色质状态,使基因偏向激活或抑制,从而改变谱系结果 158 、 159 、 160 。为了检测这种现象,开发多视角学习方法至关重要。为了填补这一空白,OT 允许整合配对的多组学数据 161 和跨模态推断 23 、 24 。最后,OT 还可用于图谱创建、批量校正 47、162 或包含单个或多个数据模态(例如,单细胞基因组学和数字病理学 163 )的患者队列 164、165、166、167 的数据集的多尺度集成。这揭示了患者 168 多尺度表征之间的结构和异质性以及相似性。
Spatial reconstruction of tissues
组织空间重建
Although the growing availability of spatial omics technologies provides tools to decipher tissue composition, leveraging and spatially reconstructing the vast amount of already available single-cell data are key to enhancing our current understanding. Furthermore, many spatial transcriptomics methods lack single-cell resolution (such as Visium, Slide-seq169 and so on) or cover only partial transcriptomes (such as MERFISH170, SeqFISH171 and so on) and require the integration of scRNA-seq data to provide a holistic understanding of the spatial organization of in-depth cellular states within tissues.
尽管空间组学技术的日益普及为解读组织组成提供了工具,但利用和空间重建大量现有的单细胞数据才是增强我们当前理解的关键。此外,许多空间转录组学方法缺乏单细胞分辨率(例如 Visium、Slide-seq 169 等)或仅覆盖部分转录组(例如 MERFISH 170 、SeqFISH 171 等),需要整合 scRNA-seq 数据才能全面理解组织内细胞状态的空间组织结构。
We introduced earlier how OT can be applied to reconstruct or decipher tissue composition: using the Gromov–Wasserstein plan to construct a gene expression cartography by mapping single-cell expression profiles to a reference spatial atlas19,20 (Example 3), or using the fused Gromov–Wasserstein plan to pairwise align spatial transcriptomics slides112, facilitating the discovery of multicellular communities or niches.
我们之前介绍了如何应用 OT 来重建或解读组织组成:使用 Gromov–Wasserstein 计划通过将单细胞表达谱映射到参考空间图谱 19、20 (示例 3)来构建基因表达制图,或使用融合的 Gromov–Wasserstein 计划成对比对空间转录组学幻灯片 112 ,以促进多细胞群落或生态位的发现。
In addition, OT has been utilized to determine clusters of different cell types across tissues, to provide a coarse-grained understanding of the spatial tissue architecture. For example, a novel optimization framework based on the Gromov–Wasserstein distance for fast cell-type decomposition of spatial omics has been proposed172. Another method infers spatial and signalling relationships between cells from single-cell transcriptomic data by relying on structured OT91,173. This approach constructs a spatial metric for cells in scRNA-seq data to reconstruct cell–cell communication networks and identify intercellular regulatory relationships between genes. A further extension of this approach infers cell–cell communication in spatial transcriptomics data by integrating biochemical signalling through ligand–receptor binding92. The collective OT method thereby handles complex molecular interactions and spatial constraints by accounting for the competition between different ligand and receptor species and spatial distances between cells.
此外,OT 已用于确定组织中不同细胞类型的聚类,以提供对空间组织结构的粗粒度理解。例如,已提出了一种基于 Gromov-Wasserstein 距离的新型优化框架,用于快速分解空间组学的细胞类型 172 。另一种方法是依靠结构化的 OT 91,173 从单细胞转录组数据中推断细胞之间的空间和信号传导关系。该方法为 scRNA-seq 数据中的细胞构建了空间度量,以重建细胞间通讯网络并识别基因之间的细胞间调控关系。该方法的进一步扩展通过整合通过配体-受体结合的生化信号来推断空间转录组学数据中的细胞间通讯 92 。因此,集体 OT 方法通过考虑不同配体和受体种类之间的竞争以及细胞之间的空间距离来处理复杂的分子相互作用和空间约束。
Reproducibility and data deposition
可重复性和数据存储
All concepts, methods and algorithms introduced in this Primer are accompanied by open-source data sets and readily available libraries.
本入门书中介绍的所有概念、方法和算法都附带开源数据集和现成的库。
Various open-source libraries implement the OT methods presented here and can be integrated into different research workflows. Notably, libraries such as the Optimal Transport Toolbox (OTT)26 based on Python’s JAX library174, the Python Optimal Transport (POT)175 package integrating both NumPy176 and PyTorch177 implementations and the GeomLoss178 library based on PyTorch offer implementations of OT algorithms, metrics and visualization techniques. These libraries lower entry barriers and enable researchers to easily apply and reproduce analyses across data sets. In addition, OTT implements different neural OT solvers. Finally, running the OTT library at its core, the Multi-Omics Single-Cell Optimal Transport (moscOT)68 Python package provides methods as well as tutorials for using OT in single-cell and spatial omics. moscOT covers many applications highlighted in this Primer, including OT for reconstructing cell differentiation processes, with extensions to incorporate lineage information and capturing spatiotemporal dynamics, multimodal omics integration as well as spatial reconstruction of tissues.
各种开源库都实现了这里介绍的 OT 方法,并且可以集成到不同的研究工作流程中。值得注意的是,基于 Python 的 JAX 库 174 的最佳传输工具箱 (OTT) 26 、集成 NumPy 176 和 PyTorch 177 实现的 Python 最佳传输 (POT) 175 包以及基于 PyTorch 的 GeomLoss 178 库等库提供了 OT 算法、指标和可视化技术的实现。这些库降低了进入门槛,使研究人员能够轻松地跨数据集应用和重现分析。此外, OTT 实现了不同的神经 OT 求解器。最后,以 OTT 库为核心的多组学单细胞最佳传输 (moscOT) 68 Python 包提供了在单细胞和空间组学中使用 OT 的方法和教程。 moscOT 涵盖了本入门指南中重点介绍的许多应用,包括用于重建细胞分化过程的 OT,以及合并谱系信息和捕获时空动态、多模态组学整合以及组织空间重建的扩展。
Many large-scale single-cell and spatial omics data sets are openly accessible, allowing researchers to evaluate and validate new computational methods and algorithms. Open-access data sets allow to compare or benchmark novel algorithms against established methods, enabling a fair and standardized evaluation of their performance. Examples relevant to this Primer include data for studying the reconstruction of developmental processes5,127, spatiotemporal analysis179 and incorporation of lineage information180,181,182,183; drug perturbation effects6,73,184, including across cells from different patients185,186; multimodal data integration187,188; analysis of cellular MEs189; identification of cell states190,191; and spatial reconstruction of tissues20 or alignment of spatial transcriptomics data112, besides major single-cell data set collections such as the Human Cell Atlas192 and the Cell×Gene database193,194. For further information and access to the processed data sets, we refer the reader to recent benchmark papers6,121,195 and libraries such as moscOT68.
许多大规模单细胞和空间组学数据集都是开放的,允许研究人员评估和验证新的计算方法和算法。开放获取数据集允许将新算法与已建立的方法进行比较或基准测试,从而能够公平、标准化地评估它们的性能。与本入门书相关的例子包括用于研究重建发育过程 5、127 、 时空分析 179 和整合谱系信息 180、181、182、183 的数据;药物扰动效应 6、73、184 , 包括跨不同患者的细胞 185、186 ; 多模态数据整合 187、188 ;细胞 ME 分析 189 ;细胞状态识别 190、191 ; 以及组织的空间重建 20 或空间转录组学数据的比对 112 ,此外还有主要的单细胞数据集集合,例如人类细胞图谱 192 和细胞×基因数据库 193 , 194 。有关更多信息和访问处理后的数据集,我们请读者参阅最近的基准论文 6 、 121 、 195 和 moscOT 68 等库。
When publishing an OT analysis, reasonable minimum reporting standards include clearly formulating the optimization problem solved and providing code and data to reproduce numerical results.
发布 OT 分析时,合理的最低报告标准包括清晰地表述所解决的优化问题并提供代码和数据以重现数值结果。
Limitations and optimizations
限制和优化
Computational aspects 计算方面
Solving the OT problem was historically viewed as a computationally intensive endeavour, which strongly limited its applicability to large-scale data science problems. The computational approaches proposed in this Primer mostly rely on two ingredients that can bring down costs in the age of GPUs: entropy-regularized formulations that rely on the Sinkhorn algorithm196 to compute couplings, and neural approaches that can model transport maps. The current rough complexity estimate for Sinkhorn-based approaches is quadratic in the number of points, as in O(nm). As a result, most solvers will easily deal with sizes of n, m ≈ 20,000 points. Larger point clouds typically require a more careful memory management, to avoid materializing these large nm matrices26. For even larger point clouds, low-rank solvers are currently the only viable approach for n, m ≥ 500,000 points, as demonstrated in ref. 68, as their complexity is typically of the order of (n + m)r to yield a coupling of rank r, putting aside various constants that depend on the dimension of the point clouds. We do expect, however, that Sinkhorn algorithm-based methods will soon reach these scales, by streaming quadratic operations more efficiently on multi-GPU machines. Neural-network-based approaches are learned using stochastic gradient descent and scale, therefore, more efficiently to much larger sample sizes, as they do not view these point clouds as a whole, but rather as minibatches. Although this can unlock much larger-scale applications, the common downside of these approaches stems from the non-convex nature of this approach. This requires more familiarity with the intricacies of neural optimization and typically results in more variability.
解决 OT 问题历来被视为计算密集型工作,这极大地限制了其在大规模数据科学问题中的适用性。本入门书中提出的计算方法主要依赖于两个可以在 GPU 时代降低成本的要素:依赖于 Sinkhorn 算法 196 来计算耦合的熵正则化公式,以及可以建模传输图的神经方法。目前基于 Sinkhorn 的方法的复杂度粗略估计是点数的二次方,即 O ( nm )。因此,大多数求解器可以轻松处理 n , m ≈ 20,000 个点的大小。更大的点云通常需要更仔细的内存管理,以避免实现这些大的 nm 矩阵 26。 对于更大的点云,低秩求解器目前是 n , m ≥ 500,000 点的唯一可行方法,如参考文献所示。 68 ,因为它们的复杂度通常为 ( n + m ) r 量级,产生秩为 r 的耦合,撇开取决于点云维度的各种常数不谈。然而,我们确实期望基于 Sinkhorn 算法的方法能够通过在多 GPU 机器上更高效地流式执行二次运算,很快达到这些规模。基于神经网络的方法使用随机梯度下降和缩放进行学习,因此,对于更大的样本量,它们能够更有效地处理,因为它们不将这些点云视为一个整体,而是将其视为小批量。 虽然这可以解锁更大规模的应用,但这些方法的共同缺点源于其非凸性。这需要更熟悉神经优化的复杂性,并且通常会导致更大的可变性。
Modelling aspects 建模方面
All cell differentiation methods that aim to uncover single-cell trajectories from population data face a common challenge: multiple dynamics and mechanisms of action can generate the observed sequential distribution of cell states, making it necessary to make assumptions about the underlying cellular dynamics, such as continuity in time. In that sense, OT proposes a new inductive bias that makes few implicit assumptions about the nature of biological systems and processes. For example, employing OT to reconstruct cellular perturbation responses is based on the hypothesis that chemical drugs or genetic alterations incrementally and continuously change the molecular profiles of cells from the untreated state to the new perturbed state. On the contrary, if a perturbation substantially and abruptly disrupts the population structure (relative to the timescale of the measurement), or large parts of the cells undergo apoptosis, OT will likely not be able to catch these underlying cellular dynamics correctly, as the minimum effort hypothesis6 stands at its core. In these scenarios, the accuracy of OT-based methods is likely to suffer. Ultimately, fine granularity of measurements throughout time is necessary to recover large cell state changes between consecutive time points successfully.
所有旨在从群体数据中揭示单细胞轨迹的细胞分化方法都面临着一个共同的挑战:多种动力学和作用机制可以产生观察到的细胞状态的序列分布,因此有必要对潜在的细胞动力学做出假设,例如时间上的连续性。从这个意义上讲,OT 提出了一种新的归纳偏差,它几乎不做任何关于生物系统和过程本质的隐式假设。例如,使用 OT 重建细胞扰动响应是基于这样的假设:化学药物或基因改变会逐步且持续地改变细胞的分子谱,使其从未处理状态转变为新的扰动状态。相反,如果扰动显著且突然地破坏了群体结构(相对于测量的时间尺度而言),或者大量细胞发生凋亡,OT 可能无法正确捕捉这些潜在的细胞动力学,因为最小努力假设 6 是其核心。在这种情况下,基于 OT 的方法的准确性可能会受到影响。最终,需要在整个时间范围内进行细粒度的测量,才能成功恢复连续时间点之间的大型细胞状态变化。
It is worth noting, however, that alternative and OT-independent methods are equally affected by such scenarios: as these problems are ill-defined, it is challenging to identify the correct solution. In such cases, more complex mathematical tools that can incur more granular previous knowledge are required. Tools, such as causal frameworks197 or mechanistic models103,198, however, are often not able to scale to settings with thousands of involved components — such as with large-scale gene regulatory networks — and in most cases the true mechanisms are unknown.
然而,值得注意的是,替代方法和独立于 OT 的方法同样会受到此类场景的影响:由于这些问题定义不明确,因此很难找到正确的解决方案。在这种情况下,需要更复杂的数学工具来获取更精细的先前知识。然而,诸如因果框架 197 或机制模型 103、198 之类的工具通常无法扩展到包含数千个组件的场景(例如大规模基因调控网络),而且在大多数情况下,真正的机制尚不清楚。
When reconstructing cellular dynamics, OT-based methods may not be able to recover complex dynamics characterized by rotations and oscillations between consecutive snapshots if these dynamics are not captured by measurements180. Informed choices of cost functions that, for example, integrate previous knowledge of the underlying mechanisms might address this limitation. OT methods for single-cell and spatial genomics often use the Euclidean distance as a cost function, owing to its theoretical properties and practicality. However, these distances may become less discriminative in high-dimensional spaces. As a result, the OT problem is often cast into a lower-dimensional representation of the data14. Although metric learning approaches could alleviate this issue90,199, the adaptive selection of robust cost functions remains a crucial area for future research.
重建细胞动力学时,如果测量结果无法捕捉到细胞动态,基于 OT 的方法可能无法恢复连续快照之间以旋转和振荡为特征的复杂动态 180 。明智地选择成本函数(例如,整合先前对底层机制的了解)可能会解决这一限制。单细胞和空间基因组学的 OT 方法通常使用欧几里得距离作为成本函数,因为它具有理论特性和实用性。然而,这些距离在高维空间中可能变得不那么具有辨别力。因此,OT 问题通常被转化为数据的低维表示 14 。虽然度量学习方法可以缓解这个问题 90,199 ,但自适应地选择稳健的成本函数仍然是未来研究的关键领域。
Finally, non-measured cell features such as epigenetic states may influence the overall dynamics. Any algorithm inferred from data, however, will only uncover phenomena captured by the provided data modality.
最后,表观遗传状态等未测量的细胞特征可能会影响整体动态。然而,任何基于数据推断的算法都只能揭示特定数据模态所捕获的现象。
The application of trajectory inference and perturbation response prediction are not the only settings relying on certain hypothesis to apply OT. In the application of multimodality integration, the ground-truth correspondence between measurement readouts of different high-throughput technologies is, in most cases, unknown. OT solutions align different modalities based on similarities in the structure of each data set, such as by comparing the pairwise similarities between individual cells of each modality. This, however, relies on the hypothesis that measurements resulting from different modalities are all manifestations of the same distribution of cell states. Finally, using OT to reconstruct tissues or align spatial omics data relies on the hypothesis that similar cell states lie in physical proximity. Strong deviations from these hypotheses ultimately affect the quality of the computational result achieved through applying various OT formulations.
轨迹推断和扰动响应预测的应用并不是依赖某些假设来应用 OT 的唯一设置。在多模态整合的应用中,不同高通量技术的测量读数之间的真实对应关系在大多数情况下是未知的。OT 解决方案根据每个数据集结构的相似性来对齐不同的模态,例如通过比较每个模态中各个细胞之间的成对相似性。然而,这依赖于这样的假设:不同模态产生的测量结果都是相同细胞状态分布的表现。最后,使用 OT 重建组织或对齐空间组学数据依赖于相似的细胞状态位于物理接近性的假设。与这些假设的强烈偏差最终会影响通过应用各种 OT 公式获得的计算结果的质量。
Outlook 前景
The inherently complex and constantly changing interactions in biological systems call for innovative computational approaches. With its static and dynamic formulations and recent deep learning developments, OT has provided an indispensable framework for high-throughput, multimodal and multiscale molecular, cell, tissue and organ biology. We anticipate that the increasing data complexity across multiple levels of biological organization, from molecular and cellular through spatial profiling of tissues and imaging of organs, will further cement that status. As we look ahead, several key questions emerge that shape the future of this research.
生物系统中固有的复杂且不断变化的相互作用需要创新的计算方法。凭借其静态和动态公式以及近期深度学习的发展,OT 为高通量、多模态和多尺度的分子、细胞、组织和器官生物学提供了不可或缺的框架。我们预计,从分子和细胞到组织的空间分析和器官成像,生物组织多个层面的数据复杂性日益增加,将进一步巩固这一地位。展望未来,一些关键问题将决定该研究的未来。
First, there are novel challenges for theoretical and algorithmic development within the OT field. As reviewed in this Primer, beyond mappings and couplings, OT provides a mathematical link to geometric variational frameworks that allow studying flows of distributions on metric spaces43,128,129,139,143. This enables us to model cellular dynamics as gradient flows9,13 or optimal control problems described through systems of SDEs11,62,69,134. These concepts coincide with active research in the field of deep learning concerned with robust parameterizations of flows109,133,200,201,202,203,204 and dynamical systems11,69,130,131,205,206,207. In particular, recent deep learning parameterizations of dynamic OT contain technologies known as diffusion generative models208,209 and flow matching methods109,200,202, an emerging generative model class that has achieved remarkable results in synthesizing high-fidelity data210,211. These advancements arise as a response to the distinctive challenges and characteristics presented by single-cell and spatial biology, underscoring how progress in high-throughput biology catalyses the creation of innovative methodological designs, algorithmic principles and novel concepts within the artificial intelligence community. For example, when aiming at enriching single-cell data with spatial information of tissue pathology data, histological information extracted from haematoxylin-and-eosin stains must be integrated with single-cell data212. The ability of OT to translate between multiple modalities can be used as a starting point for machine learning algorithms that can generate the spatially resolved single-cell omics data set of a tissue sample from its haematoxylin-and-eosin histology image.
首先,OT 领域的理论和算法发展面临着新的挑战。正如本入门书所回顾的,除了映射和耦合之外,OT 还提供了与几何变分框架的数学联系,这些框架可以研究度量空间上的分布流 43、128、129、139、143 。 这使我们能够将细胞动力学建模为梯度流 9、13 或通过 SDE 系统描述的最优控制问题 11、62、69、134 。 这些概念与深度学习领域中涉及流的稳健参数化 109、133、200、201、202、203、204 和动力系统 11、69、130、131、205、206、207 的活跃研究相一致 。 具体来说,动态 OT 的最新深度学习参数化包含称为扩散生成模型 208、209 和流匹配方法 109、200、202 的技术,这是一种新兴的生成模型类,在合成高保真数据 210、211 方面取得了显著成果。 这些进步是对单细胞和空间生物学所提出的独特挑战和特点的回应,强调了高通量生物学的进步如何催化人工智能社区内创新方法设计、算法原理和新概念的产生。例如,当旨在利用组织病理数据的空间信息丰富单细胞数据时,必须将从苏木精和伊红染色中提取的组织学信息与单细胞数据 212 相结合。OT 在多种模态之间转换的能力可以作为机器学习算法的起点,该算法可以从组织样本的苏木精和伊红组织学图像生成空间分辨的单细胞组学数据集。
With the rise of single-cell foundation models213,214 as well as the vision of an artificial-intelligence-powered virtual cell integrating large biomedical data sets into a universal representation of cells across modalities, tissues and species, the role of OT will be once more redefined: as a building block and as a connecting unit of connected deep learning models that allow us to simulate cell behaviour, translate across different temporal and physical scales and connect different measurements.
随着单细胞基础模型 213、214 的兴起,以及由人工智能驱动的虚拟细胞将大型生物医学数据集整合为跨模态、组织和物种的细胞通用表示的愿景,OT 的作用将再次被重新定义:作为构建块和连接的深度学习模型的连接单元,使我们能够模拟细胞行为,跨不同的时间和物理尺度进行转换,并连接不同的测量值。
By contrast, concurrent developments in biological experiments continuously aim at overcoming the technological limitation of destructive cell assays: besides existing live imaging approaches110,215, Chen et al.108, for example, propose a transcriptome profiling approach that preserves cell viability. Cell differentiation processes have been captured by clonally connecting cells and their progenitors through barcodes107. These methods thus offer (lower-throughput) insights that provide individual trajectories of cells over time by aligning between distinct measurement snapshots. To align OT algorithms with advances on the experimental side, novel algorithmic frameworks need to be developed, which make use of such (partially) aligned data sets109,133,204,216.
相比之下,生物实验的同步发展不断致力于克服破坏性细胞测定的技术限制:除了现有的活体成像方法 110、215 之外 ,例如,Chen 等人 108 提出了一种保留细胞活力的转录组分析方法。通过条形码 107 克隆连接细胞及其祖细胞,可以捕获细胞分化过程。因此,这些方法通过对齐不同的测量快照,提供(低通量)洞察,提供细胞随时间的个体轨迹。为了使 OT 算法与实验方面的进展保持一致,需要开发新的算法框架,利用这种(部分)对齐的数据集 109、133、204、216 。
Although the neural OT methods highlighted here show promise in modelling perturbation responses and demonstrate the versatility and out-of-sample generalization capacities in various applications, their performance has only been evaluated on relatively small data sets so far185,186. It is evident that approaches addressing these challenges could readily exploit the upcoming availability of large-scale patient cohort studies, comprising individuals with distinct molecular profiles217, in vivo screens of complex tissues218 and large-scale perturbation screens of combinatorial libraries219,220,221.
尽管这里强调的神经 OT 方法在建模扰动响应方面显示出良好的前景,并在各种应用中展示了多功能性和样本外泛化能力,但它们的性能迄今为止仅在相对较小的数据集上进行了评估 185 , 186 。显然,解决这些挑战的方法可以很容易地利用即将到来的大规模患者队列研究,包括具有不同分子谱的个体 217 、复杂组织的体内筛选 218 和组合库的大规模扰动筛选 219 、 220 、 221 。
Although single-cell and spatial omics give insights into the molecular composition of cells and tissues, cellular decision-making might be influenced by other confounding factors, difficult to capture using existing technologies. The returned OT plan or map, however, is not calibrated in terms of confidence in the prediction or uncertainty in the output of the model. The rise of personalized medicine and the growing importance of artificial-intelligence-based clinical decision-making calls for algorithmic solutions that account for such confounding factors and augment existing systems to integrate confidence measures. By providing probabilistic assignments between data samples, OT has the capacity to achieve this. With its rich properties, astonishing mathematical connections and innovative numerical implementations26, OT thus makes an exciting avenue of future work to facilitate novel biological discoveries, infer personalized therapies from single-cell patient samples and push the boundaries of regenerative medicine.
尽管单细胞和空间组学可以深入了解细胞和组织的分子组成,但细胞决策可能受到其他混杂因素的影响,而这些因素很难用现有技术捕捉到。但是,返回的 OT 计划或地图并未根据预测的置信度或模型输出的不确定性进行校准。个性化医疗的兴起和基于人工智能的临床决策日益重要,要求算法解决方案能够考虑这些混杂因素并增强现有系统以整合置信度指标。通过提供数据样本之间的概率分配,OT 有能力实现这一点。凭借其丰富的特性、惊人的数学联系和创新的数值实现 26 ,OT 为未来工作开辟了一条令人兴奋的途径,以促进新的生物学发现,从单细胞患者样本中推断个性化疗法,并突破再生医学的界限。
References
Villani, C. Topics in Optimal Transportation Vol. 58 (American Mathematical Society, 2003).
Santambrogio, F. Optimal transport for applied mathematicians. Birkhäuser 55, 94 (2015).
Figalli, A. The Monge–Ampère Equation and Its Applications (Zurich Lectures in Advanced Mathematics, 2017).
Caffarelli, L. A. in Optimal Transportation and Applications. Lecture Notes in Mathematics Vol. 1813 (Springer, 2003).
Schiebinger, G. et al. Optimal-transport analysis of single-cell gene expression identifies developmental trajectories in reprogramming. Cell 176, 928–943.e22 (2019). By computing consecutive optimal transport plans between measurement snapshots, this work reconstructs developmental processes from single-cell data.
Bunne, C. et al. Learning single-cell perturbation responses using neural optimal transport. Nat. Methods 20, 1759–1768 (2023). Using neural optimal transport based on Brenier’s theorem, this work allows us to predict the perturbation responses of heterogeneous cell populations to chemical drugs, developmental signals or genetic perturbations.
Uscidda, T. & Cuturi, M. The Monge gap: a regularizer to learn all transport maps. In Int. Conf. Machine Learning (ICML, 2023). This paper introduces the Monge gap, a regularizer for neural optimal transport (OT) methods that quantifies how far a map T deviates from the ideal properties we expect from an OT map.
Uhler, C. & Shivashankar, G. Machine learning approaches to single-cell data integration and translation. Proc. IEEE 110, 557–576 (2022).
Bunne, C., Meng-Papaxanthos, L., Krause, A. & Cuturi, M. Proximal optimal transport modeling of population dynamics. In Int. Conf. Artificial Intelligence and Statistics Vol. 25 (AISTATS, 2022). Building on the connection of optimal transport, gradient flows and partial differential equations, this work learns an energy potential that explains the continuous differentiation of single cells over time.
Forrow, A. & Schiebinger, G. LineageOT is a unified framework for lineage tracing and trajectory inference. Nat. Commun. 12, 4940 (2021).
Bunne, C., Hsieh, Y.-P., Cuturi, M. & Krause, A. The Schrödinger bridge between Gaussian measures has a closed form. In Int. Conf. Artificial Intelligence and Statistics Vol. 206 (AISTATS, 2023).
Bunne, C.、Hsieh, Y.-P.、Cuturi, M. 和 Krause, A. 高斯测度之间的薛定谔桥具有闭合形式。载于国际人工智能与统计学会议第 206 卷(AISTATS,2023 年)。Lavenant, H., Zhang, S., Kim, Y.-H. & Schiebinger, G. Towards a mathematical theory of trajectory inference. Ann. Appl. Probab. 34, 428–500 (2024).
Lavenant, H.,Zhang, S.,Kim, Y.-H. 和 Schiebinger, G. 迈向轨迹推断的数学理论。 《应用概率年鉴》 34,428–500 (2024)。Tong, A., Huang, J., Wolf, G., Van Dijk, D. & Krishnaswamy, S. TrajectoryNet: a dynamic optimal transport network for modeling cellular dynamics. In Int. Conf. Machine Learning (ICML, 2020). This paper parameterizes dynamic optimal transport and in particular the Benamou–Brenier formulation using normalizing flows and enables to generate representative single-cell trajectories from snapshot measurements.
Tong, A., Huang, J., Wolf, G., Van Dijk, D. 和 Krishnaswamy, S. TrajectoryNet:用于建模细胞动力学的动态最优传输网络。发表于国际机器学习大会 (ICML, 2020)。 本文使用归一化流参数化了动态最优传输,特别是 Benamou-Brenier 公式,并能够从快照测量中生成具有代表性的单细胞轨迹。Yang, K. D. et al. Predicting cell lineages using autoencoders and optimal transport. PLoS Comput. Biol. 16, e1007828 (2020).
Yang, KD 等人。使用自动编码器和最优传输预测细胞谱系。PLoS Comput. Biol. 16 , e1007828 (2020)。Zhang, S., Afanassiev, A., Greenstreet, L., Matsumoto, T. & Schiebinger, G. Optimal transport analysis reveals trajectories in steady-state systems. PLoS Comput. Biol. 17, e1009466 (2021).
Zhang, S., Afanassiev, A., Greenstreet, L., Matsumoto, T. 和 Schiebinger, G. 最佳传输分析揭示了稳态系统中的轨迹。PLoS Comput. Biol. 17 , e1009466 (2021)。Chizat, L., Zhang, S., Heitz, M. & Schiebinger, G. Trajectory inference via mean-field Langevin in path space. In Advances in Neural Information Processing Systems (NeurIPS, 2022).
Chizat, L.、Zhang, S.、Heitz, M. 和 Schiebinger, G. 通过路径空间中的平均场朗之万进行轨迹推断。载于《神经信息处理系统进展》 (NeurIPS,2022)。Yang, K. D. & Uhler, C. Scalable unbalanced optimal transport using generative adversarial networks. In Int. Conf. Learning Representations (ICLR, 2019).
Yang, KD & Uhler, C. 基于生成对抗网络的可扩展不平衡最优传输。发表于国际会议“学习表征” (ICLR,2019)。Lübeck, F. et al. Neural unbalanced optimal transport via cycle-consistent semi-couplings. Preprint at https://arxiv.org/abs/2209.15621 (2022).
Lübeck, F. 等人。通过循环一致半耦合实现神经非平衡最优传输。预印本网址: https://arxiv.org/abs/2209.15621 (2022)。Moriel, N. et al. NovoSpaRc: flexible spatial reconstruction of single-cell gene expression with optimal transport. Nat. Protocols 16, 4177–4200 (2021).
Moriel,N.等人。NovoSpaRc:通过最佳运输实现单细胞基因表达的灵活空间重建。 《国家议定书》 16,4177–4200 (2021)。Nitzan, M., Karaiskos, N., Friedman, N. & Rajewsky, N. Gene expression cartography. Nature 576, 132–137 (2019). This paper introduces how optimal transport across heterogeneous spaces, that is, the Gromov–Wasserstein distance, can be used to spatially reconstruct tissues or map non-spatially resolved single-cell measurement onto a reference atlas.
Nitzan, M.、Karaiskos, N.、Friedman, N. 和 Rajewsky, N. 基因表达制图。 《自然》 576 , 132–137 (2019)。 本文介绍了如何利用跨异质空间的最优传输(即 Gromov-Wasserstein 距离)在空间上重建组织或将非空间分辨的单细胞测量数据映射到参考图谱上。Demetci, P., Santorella, R., Sandstede, B., Noble, W. S. & Singh, R. SCOT: single-cell multi-omics alignment with optimal transport. J. Comput. Biol. 29, 3–18 (2022). Building on optimal transport extensions to heterogeneous spaces, this method allows to integrate and translate across multiple data modalities.
Demetci, P.、Santorella, R.、Sandstede, B.、Noble, WS 和 Singh, R. SCOT:基于最优传输的单细胞多组学比对。 《计算机生物学杂志》 29 , 3–18 (2022)。 基于最优传输扩展到异构空间,该方法可以跨多种数据模态进行集成和转换。Huizing, G.-J., Peyré, G. & Cantini, L. Optimal transport improves cell–cell similarity inference in single-cell omics data. Bioinformatics 38, 2169–2177 (2022).
Huizing, G.-J., Peyré, G. & Cantini, L. 最佳传输改进了单细胞组学数据中细胞间相似性推断。 《生物信息学》 38 , 2169–2177 (2022)。Yang, K. D. & Uhler, C. Multi-domain translation by learning uncoupled autoencoders. Preprint at https://arxiv.org/abs/1902.03515 (2019).
Yang, KD & Uhler, C. 通过学习解耦自编码器实现多领域翻译。预印本链接: https://arxiv.org/abs/1902.03515 (2019)。Alatkar, S. A. & Wang, D. CMOT: cross-modality optimal transport for multimodal inference. Genome Biol. 24, 163 (2023). The study presents cross-modality optimal transport, a computational approach that aligns multimodal single-cell sequencing data into a common latent space, effectively inferring missing modalities and enhancing biological interpretations across various applications.
Alatkar, SA & Wang, D. CMOT:用于多模态推断的跨模态最优传输。 《基因组生物学》 24 , 163 (2023)。 该研究提出了跨模态最优传输,这是一种将多模态单细胞测序数据比对到共同潜在空间的计算方法,可有效推断缺失的模态,并增强各种应用中的生物学解释。Cuturi, M. Sinkhorn distances: lightspeed computation of optimal transport. In Advances in Neural Information Processing Systems Vol. 26 (NeurIPS, 2013).
Cuturi, M. Sinkhorn 距离:最优传输的光速计算。载于《神经信息处理系统进展 》第 26 卷(NeurIPS,2013)。Cuturi, M. et al. Optimal Transport Tools (OTT): A JAX Toolbox for all things Wasserstein. Preprint at https://arxiv.org/abs/2201.12324; https://github.com/ott-jax/ott (2022). A Python library build on JAX to compute optimal transport (OT) at scale, also providing implementations of various neural-network-based OT approaches.
Cuturi, M. 等人。最优传输工具 (OTT):一个适用于 Wasserstein 的 JAX 工具箱。预印本链接: https://arxiv.org/abs/2201.12324 ; https://github.com/ott-jax/ott (2022)。 这是一个基于 JAX 构建的 Python 库,用于大规模计算最优传输 (OT),并提供各种基于神经网络的 OT 方法的实现。Bunne, C., Krause, A. & Cuturi, M. Supervised training of conditional Monge maps. In Advances in Neural Information Processing Systems Vol. 35 (NeurIPS, 2022).
Bunne, C.、Krause, A. 和 Cuturi, M. 条件 Monge 图的监督训练。载于《神经信息处理系统进展 》第 35 卷(NeurIPS,2022 年)。Peyré, G. & Cuturi, M. in Foundations and Trends in Machine Learning Vol. 11 (Now Publishers, Inc., 2019). A book introducing in-depth optimal transport concepts and algorithms, with a particular focus on computational aspects.
Peyré, G. 和 Cuturi, M. 合著, 《机器学习基础与趋势 》第 11 卷(Now Publishers, Inc.,2019 年出版)。 本书深入介绍了最优传输的概念和算法,尤其侧重于计算方面。Cai, S., Georgakilas, G. K., Johnson, J. L. & Vahedi, G. A cosine similarity-based method to infer variability of chromatin accessibility at the single-cell level. Front. Genet. 9, 319 (2018).
Cai, S., Georgakilas, GK, Johnson, JL 和 Vahedi, G. 一种基于余弦相似度推断单细胞水平染色质可及性变异性的方法。Front . Genet. 9 , 319 (2018)。Watson, E. R., Mora, A., Taherian Fard, A. & Mar, J. C. How does the structure of data impact cell–cell similarity? Evaluating how structural properties influence the performance of proximity metrics in single cell RNA-seq data. Brief. Bioinform. 23, bbac387 (2022).
Watson, ER, Mora, A., Taherian Fard, A. & Mar, JC. 数据结构如何影响细胞间相似性?评估结构特性如何影响单细胞 RNA 测序数据中邻近性指标的表现。 简报。《生物信息学》 。23 ,bbac387 (2022)。Cuturi, M., Klein, M. & Ablin, P. Monge, Bregman and Occam: interpretable optimal transport in high-dimensions with feature-sparse maps. In Proc. 40th Int. Con. Mach. Learn. Vol. 202, 6671–6682 (PMLR, 2023).
Cuturi, M.、Klein, M. 和 Ablin, P. Monge, Bregman 和 Occam:具有特征稀疏映射的高维可解释最优传输。刊于第 40 届国际计算机学会会刊 (Proc. 40th Int. Con. Mach. Learn .),第 202 卷,第 6671–6682 页 (PMLR,2023 年)。Liu, R., Balsubramani, A. & Zou, J. Learning transport cost from subset correspondence. In Int. Conf. Learning Representations (ICLR, 2020).
Liu, R.、Balsubramani, A. 和 Zou, J. 从子集对应关系学习传输成本。发表于国际会议“学习表征” (ICLR,2020)。Stuart, A. M. & Wolfram, M.-T. Inverse optimal transport. SIAM J. Appl. Math. 80, 19M1261122 (2020).
Stuart, AM & Wolfram, M.-T. 逆最优传输。SIAM J. Appl. Math. 80 , 19M1261122 (2020)。Li, R., Ye, X., Zhou, H. & Zha, H. Learning to match via inverse optimal transport. J. Mach. Learn. Res. 20, 1–37 (2019).
Li, R., Ye, X., Zhou, H. & Zha, H. 通过逆最优传输学习匹配。 《机器学习研究》 2019 年第 20 卷 ,第 1-37 页。Monge, G. Mémoire sur la théorie des déblais et des remblais (Histoire de l’Académie Royale des Sciences, 1781).
Monge,G. 《关于挖方和堤岸理论的回忆录》 (皇家科学院历史,1781 年)。Kantorovich, L. On the transfer of masses [Russian]. In Dokl. Akad. Nauk SSSR 37, 227–229 (1942).
Kantorovich,L. 论群众的转移 [俄语]。 载于《Dokl. Akad. Nauk SSSR》 第 37 卷 ,第 227–229 页 (1942 年)。Kuhn, H. W. The Hungarian method for the assignment problem. Nav. Res. Logist. Q. 2, 83–97 (1955).
Kuhn, HW.《分配问题的匈牙利方法》。 《导航研究与逻辑学》第 2 卷 ,83–97 (1955)。Dantzig, G. Linear Programming and Extensions (Princeton Univ. Press, 1963).
Hitchcock, F. L. The distribution of a product from several sources to numerous localities. J. Math. Phys. 20, 224–230 (1941).
Koopmans, T. C. Optimum utilization of the transportation system. Econ. J. Econ. Soc. 17, 136–146 (1949).
Koopmans, TC,《交通系统的最佳利用》。 《Econ. J. Econ. Soc.》 17,136–146 (1949)。Ahuja, R. K., Magnanti, T. L. & Orlin, J. B. Network Flows: Theory, Algorithms, and Applications (Prentice Hall, 1993).
Ahuja, RK、Magnanti, TL 和 Orlin, JB, 《网络流:理论、算法和应用》 (Prentice Hall,1993 年)。Bertsekas, D. P. The auction algorithm: a distributed relaxation method for the assignment problem. Ann. Oper. Res. 14, 105–123 (1988).
Bertsekas, DP. 拍卖算法:一种用于分配问题的分布式松弛方法。Ann . Oper. Res. 14 , 105–123 (1988)。Benamou, J.-D. & Brenier, Y. A computational fluid mechanics solution to the Monge–Kantorovich mass transfer problem. Numer. Math. 84, 375–393 (2000).
Benamou, J.-D. & Brenier, Y. Monge–Kantorovich 质量传递问题的计算流体力学解。 数值数学 84 , 375–393 (2000)。Brenier, Y. Polar factorization and monotone rearrangement of vector-valued functions. Commun. Pure Appl. Math. 44, 375–417 (1991).
Brenier,Y. 向量值函数的极分解和单调重排。 《纯粹应用数学通讯》 44,375–417 (1991)。Scetbon, M., Cuturi, M. & Peyré, G. Low-rank Sinkhorn factorization. In Int. Conf. Machine Learning Vol. 139 (ICML, 2021).
Scetbon, M.、Cuturi, M. 和 Peyré, G. 低秩 Sinkhorn 分解。载于国际机器学习会议第 139 卷 (ICML, 2021)。Scetbon, M. & Cuturi, M. Low-rank optimal transport: approximation, statistics and debiasing. In Advances in Neural Information Processing Systems (NeurIPS) Vol. 35 (NeurIPS, 2022).
Scetbon, M. & Cuturi, M. 低秩最优传输:近似、统计与去偏。载于《神经信息处理系统进展》 ( NeurIPS )第 35 卷(NeurIPS,2022 年)。Forrow, A. et al. Statistical optimal transport via factored couplings. In Int. Conf. Artificial Intelligence and Statistics (AISTATS) 2454–2465 (PMLR, 2019).
Forrow, A. 等人。通过因子耦合实现统计最优传输。发表于国际人工智能与统计学会议 ( AISTATS )第 2454–2465 页(PMLR,2019 年)。Dudley, R. M. et al. Weak convergence of probabilities on nonseparable metric spaces and empirical measures on Euclidean spaces. Ill. J. Math. 10, 109–126 (1966).
Dudley,RM 等人。不可分度量空间上概率的弱收敛与欧氏空间上的经验测度。 《伊利诺伊州数学杂志》 10,109–126 (1966)。Boissard, E. & Le Gouic, T. On the mean speed of convergence of empirical and occupation measures in Wasserstein distance. Annales de l’IHP Probabilités et statistiques 50, 539–563 (2014).
Boissard, E. & Le Gouic, T. 关于 Wasserstein 距离中经验测量和占领测量的收敛平均速度。 年鉴 de l'IHP 概率与统计 50 , 539–563 (2014)。Pooladian, A.-A. & Niles-Weed, J. Entropic estimation of optimal transport maps. Preprint at https://arxiv.org/abs/2109.12004 (2021).
Pooladian, A.-A. & Niles-Weed, J. 最优传输图的熵估计。预印本链接: https://arxiv.org/abs/2109.12004 (2021)。Finlay, C., Gerolin, A., Oberman, A. M. & Pooladian, A.-A. Learning normalizing flows from entropy-Kantorovich potentials. Preprint at https://arxiv.org/abs/2006.06033 (2020).
Finlay, C.、Gerolin, A.、Oberman, AM 和 Pooladian, A.-A. 从熵-Kantorovich 势学习正则化流。预印本网址: https://arxiv.org/abs/2006.06033 (2020)。Wilfrid, G. & Robert, J. M. The geometry of optimal transportation. Acta Math. 177, 113–161 (1996).
Goodfellow, I. et al. Generative adversarial networks. In Advances in Neural Information Processing Systems Vol. 63 (NeurIPS, 2014).
Arjovsky, M., Chintala, S. & Bottou, L. Wasserstein generative adversarial networks. In Int. Conf. Machine Learning (ICML, 2017).
Chen, R. T., Rubanova, Y., Bettencourt, J. & Duvenaud, D. Neural ordinary differential equations. In Advances in Neural Information Processing Systems (NeurIPS, 2018).
Makkuva, A., Taghvaei, A., Oh, S. & Lee, J. Optimal transport mapping via input convex neural networks. In Int. Conf. Machine Learning Vol. 119 (ICML, 2020).
Korotin, A., Egiazarian, V., Asadulaev, A., Safin, A. & Burnaev, E. Wasserstein-2 generative networks. Preprint at https://arxiv.org/abs/1909.13082 (2019).
Alvarez-Melis, D., Schiff, Y. & Mroueh, Y. Optimizing functionals on the space of probabilities with input convex neural networks. Trans. Mach. Learn. Res. (2022).
Mokrov, P. et al. Large-scale Wasserstein gradient flows. In Advances in Neural Information Processing Systems (NeurIPS, 2021).
Vaswani, A. et al. Attention is all you need. In Advances in Neural Information Processing Systems (NeurIPS, 2017).
Eyring, L. V. et al. Modeling single-cell dynamics using unbalanced parameterized Monge maps. In International Conference on Learning Representations (ICLR, 2024).
Pariset, M., Hsieh, Y.-P., Bunne, C., Krause, A. & De Bortoli, V. Unbalanced diffusion Schrödinger bridge. Preprint at https://arxiv.org/abs/2306.09099 (2023).
Frogner, C., Zhang, C., Mobahi, H., Araya, M. & Poggio, T. A. Learning with a Wasserstein loss. In Advances in Neural Information Processing Systems Vol. 28 (NeurIPS, 2015).
Chizat, L., Peyré, G., Schmitzer, B. & Vialard, F.-X. Scaling algorithms for unbalanced optimal transport problems. Math. Comput. 87, 2563–2609 (2018).
Mémoli, F. Gromov–Wasserstein distances and the metric approach to object matching. Found. Comput. Math. 11, 417–487 (2011).
Peyré, G., Cuturi, M. & Solomon, J. Gromov–Wasserstein averaging of kernel and distance matrices. In Int. Conf. Machine Learning (ICML, 2016).
Scetbon, M., Peyré, G. & Cuturi, M. Linear-time Gromov–Wasserstein distances using low rank couplings and costs. In Int. Conf. Machine Learning Vol. 162 (ICML, 2022).
Klein, D. et al. Mapping cells through time and space with moscot. Preprint at bioRxiv https://doi.org/10.1101/2023.05.11.540374 (2023). A Python library based on Optimal Transport Toolbox that implements representative optimal transport applications in single-cell genomics in JAX.
Klein, D. 等人。利用 moscot 绘制细胞的时空图。bioRxiv 预印本 https://doi.org/10.1101/2023.05.11.540374 ( 2023)。 一个基于 Optimal Transport Toolbox 的 Python 库,可在 JAX 中实现单细胞基因组学中具有代表性的最优传输应用。Vargas, F., Thodoroff, P., Lawrence, N. D. & Lamacraft, A. Solving Schrödinger bridges via maximum likelihood. Entropy 23, 1134 (2021).
Vargas, F., Thodoroff, P., Lawrence, ND & Lamacraft, A. 通过最大似然法求解薛定谔桥。 《熵》 23,1134 (2021)。Ji, Y. et al. Optimal distance metrics for single-cell RNA-seq populations. Preprint at bioRxiv https://doi.org/10.1101/2023.12.26.572833 (2023).
Ji, Y. 等人 。单细胞 RNA 测序群体的最佳距离度量。bioRxiv 预印本 https://doi.org/10.1101/2023.12.26.572833 ( 2023)。Dixit, A. et al. Perturb-Seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866.e17 (2016).
Dixit, A. 等人。Perturb-Seq:利用可扩展的单细胞 RNA 分析技术解析混合遗传筛选中的分子回路。Cell 167 , 1853–1866.e17 (2016)。Replogle, J. M. et al. Mapping information-rich genotype–phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28 (2022).
Replogle, JM 等人。利用基因组规模的 Perturb-seq 绘制信息丰富的基因型-表型图谱。Cell 185 , 2559–2575.e28 (2022)。Srivatsan, S. R. et al. Massively multiplex chemical transcriptomics at single-cell resolution. Science. 367, 45–51 (2020).
Srivatsan, SR 等人。单细胞分辨率大规模多重化学转录组学。 《科学》 。367 , 45–51 (2020)。Norman, T. M. et al. Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 365, 786–793 (2019).
Norman, TM 等人。探索由丰富的单细胞表型构建的遗传相互作用流形。Science 365 , 786–793 (2019)。Gut, G., Herrmann, M. D. & Pelkmans, L. Multiplexed protein maps link subcellular organization to cellular states. Science 361, eaar7042 (2018).
Gut, G., Herrmann, MD & Pelkmans, L. 多重蛋白质图谱将亚细胞组织与细胞状态联系起来。Science 361 , eaar7042 (2018 ) 。Chen, W. S. et al. Uncovering axes of variation among single-cell cancer specimens. Nat. Methods 17, 302–310 (2020).
Chen, WS 等人。揭示单细胞癌症样本的变异轴。 《自然-方法》 17 , 302–310 (2020)。Lähnemann, D. et al. Eleven grand challenges in single-cell data science. Genome Biol. 21, 31 (2020).
Lähnemann, D. 等人。单细胞数据科学的十一大挑战。 《基因组生物学》 21 , 31 (2020)。Rood, J. E., Maartens, A., Hupalowska, A., Teichmann, S. A. & Regev, A. Impact of the Human Cell Atlas on medicine. Nat. Med. 8, 2486–2496 (2022).
Rood, JE、Maartens, A.、Hupalowska, A.、Teichmann, SA 和 Regev, A. 人类细胞图谱对医学的影响。 纳特。医学。 8,2486–2496 (2022)。Blondel, V. D., Guillaume, J.-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks. J. Statist. Mech. Theory Exp. 2008, P10008 (2008).
Blondel, VD, Guillaume, J.-L., Lambiotte, R. 和 Lefebvre, E. 大型网络中社区的快速展开。 《统计力学理论与实验杂志》 2008 ,P10008(2008)。Duò, A., Robinson, M. D. & Soneson, C. A systematic performance evaluation of clustering methods for single-cell RNA-seq data. F1000 Res. 7, 1141 (2018).
Duò, A., Robinson, MD & Soneson, C. 单细胞 RNA 测序数据聚类方法的系统性能评估。F1000 Res. 7 , 1141 (2018)。Traag, V. A., Waltman, L. & Van Eck, N. J. From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep. 9, 5233 (2019).
Traag, VA、Waltman, L. 和 Van Eck, NJ,《从鲁汶到莱顿:保障社区互联互通》。 《科学报告》 9 , 5233 (2019)。Wolf, F. A. et al. PAGA: graph abstraction reconciles clustering with trajectory inference through a topology preserving map of single cells. Genome Biol. 20, 59 (2019).
Wolf, FA 等人。PAGA:图抽象通过保留拓扑的单细胞图谱协调聚类与轨迹推断。 《基因组生物学》 20 , 59 (2019)。Weber, L. M. & Robinson, M. D. Comparison of clustering methods for high-dimensional single-cell flow and mass cytometry data. Cytometry Pt A 89, 1084–1096 (2016).
Weber, LM & Robinson, MD,高维单细胞流式和质谱流式数据聚类方法比较。Cytometry Pt A 89 , 1084–1096 (2016)。Wagner, A., Regev, A. & Yosef, N. Revealing the vectors of cellular identity with single-cell genomics. Nat. Biotechnol. 34, 1145–1160 (2016).
Wagner, A., Regev, A. & Yosef, N. Revealing the vectors of cellularidentity with single-cell genomics. Nat. Biotechnol. 34 , 1145–1160 (2016).McInnes, L., Healy, J., Saul, N. & Großberger, L. UMAP: uniform manifold approximation and projection. J. Open Source Softw. 3, 861 (2018).
McInnes, L.、Healy, J.、Saul, N. 和 Großberger, L. UMAP:均匀流形近似与投影。 《开源软件杂志》 3 , 861 (2018)。Wilson, N. K. et al. Combined single-cell functional and gene expression analysis resolves heterogeneity within stem cell populations. Cell Stem Cell 16, 712–724 (2015).
Wilson,NK 等人。单细胞功能和基因表达分析相结合,解决了干细胞群体内的异质性问题。 《细胞干细胞》 16,712–724 (2015)。Van der Maaten, L. & Hinton, G. Visualizing data using t-SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
Van der Maaten, L. & Hinton, G. 使用 t-SNE 可视化数据。J . Mach. Learn. Res. 9,2579–2605 (2008 年)。Gaublomme, J. T. et al. Single-cell genomics unveils critical regulators of Th17 cell pathogenicity. Cell 163, 1400–1412 (2015).
Gaublomme,JT 等人。单细胞基因组学揭示了 Th17 细胞致病性的关键调节因子。Cell 163,1400–1412 (2015 年 ) 。Huizing, G.-J., Cantini, L. & Peyré, G. Unsupervised ground metric learning using Wasserstein singular vectors. In Proc. 39th Int. Conf. Mach. Learn. (ICML, 2022).
Huizing, G.-J.、Cantini, L. 和 Peyré, G. 使用 Wasserstein 奇异向量的无监督地面度量学习。 刊于第 39 届国际机器学习会议论文集。 (ICML,2022)。Dou, J. X. et al. Learning more effective cell representations efficiently. In NeurIPS Workshop on Learning Meaningful Representations of Life (LMRL) (NeurIPS, 2022).
Dou, JX 等人。高效学习更有效的细胞表征。在 NeurIPS 学习有意义的生命表征研讨会 ( LMRL ) 上 (NeurIPS, 2022)。Cang, Z. & Nie, Q. Inferring spatial and signaling relationships between cells from single cell transcriptomic data. Nat. Commun. 11, 2084 (2020). This article introduces a method using structured optimal transport to incorporate lost spatial information into single-cell RNA-sequencing data, enabling the reconstruction of spatial cellular dynamics and improved understanding of cell–cell communications across tissues.
Cang, Z. & Nie, Q. 从单细胞转录组数据推断细胞间的空间和信号传导关系。 《自然通讯》 11 , 2084 (2020)。 本文介绍了一种利用结构化最优传输将丢失的空间信息整合到单细胞 RNA 测序数据中的方法,从而能够重建细胞空间动力学,并加深对跨组织细胞间通讯的理解。Cang, Z. et al. Screening cell–cell communication in spatial transcriptomics via collective optimal transport. Nat. Methods 20, 218–228 (2023). This paper introduces a collective optimal transport method to infer cell–cell communication in spatial transcriptomics data, able to trade-off complex molecular interactions and spatial constraints.
Cang, Z. 等人。通过集体最优传输筛选空间转录组学中的细胞间通讯。 《自然-方法》 20 , 218–228 (2023)。 本文介绍了一种集体最优传输方法来推断空间转录组学数据中的细胞间通讯,该方法能够权衡复杂的分子相互作用和空间约束。Yuan, Z. et al. SOTIP is a versatile method for microenvironment modeling with spatial omics data. Nat. Commun. 13, 7330 (2022).
Yuan, Z. 等人。SOTIP 是一种利用空间组学数据进行微环境建模的多功能方法。 《自然通讯》 13 , 7330 (2022)。Sun, D., Liu, Z., Li, T., Wu, Q. & Wang, C. STRIDE: accurately decomposing and integrating spatial transcriptomics using single-cell RNA sequencing. Nucleic Acids Res. 50, e42 (2022).
Sun, D., Liu, Z., Li, T., Wu, Q. & Wang, C. STRIDE:利用单细胞 RNA 测序准确分解和整合空间转录组学。 《核酸研究》 50 , e42 (2022)。Mani, S., Haviv, D., Kunes, R. & Pe’er, D. SPOT: spatial optimal transport for analyzing cellular microenvironments. In NeurIPS Workshop on Learning Meaningful Representations of Life (LMRL) (NeurIPS, 2022).
Mani, S.、Haviv, D.、Kunes, R. 和 Pe'er, D. SPOT:用于分析细胞微环境的空间最优传输。在 NeurIPS 学习有意义的生命表征研讨会 ( LMRL )上(NeurIPS,2022)。Haviv, D. et al. The covariance environment defines cellular niches for spatial inference. Nat. Biotechnol. https://doi.org/10.1038/s41587-024-02193-4 (2024). The study introduces an optimal-transport-based tool to effectively analyse high-resolution spatial profiling data by capturing complex cellular interactions and enhancing gene expression imputation through the gene–gene covariate structure across cells in the niche.
Haviv, D. 等人。协方差环境定义细胞微环境以进行空间推断。 《自然生物技术》, https://doi.org/10.1038/s41587-024-02193-4 (2024)。 该研究引入了一种基于最优传输的工具,通过捕捉复杂的细胞相互作用,并通过微环境内跨细胞的基因-基因协变量结构增强基因表达估算,从而有效分析高分辨率空间分析数据。Nguyen, N. D. et al. Optimal transport for mapping senescent cells in spatial transcriptomics. Preprint at bioRxiv https://doi.org/10.1101/2023.08.16.553591 (2023).
Mages, S. et al. TACCO unifies annotation transfer and decomposition of cell identities for single-cell and spatial omics. Nat. Biotechnol. 41, 1465–1473 (2023).
Held, M. et al. CellCognition: time-resolved phenotype annotation in high-throughput live cell imaging. Nat. Methods 7, 747–754 (2010).
Briggs, J. A. et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 360, eaar5780 (2018).
Mittnenzweig, M. et al. A single-embryo, single-cell time-resolved model for mouse gastrulation. Cell 184, 2825–2842.e22 (2021).
Farrell, J. A. et al. Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science 360, eaar3131 (2018).
Raue, A. et al. Data2dynamics: a modeling environment tailored to parameter estimation in dynamical systems. Bioinformatics 31, 3558–3560 (2015).
Ding, J. et al. Reconstructing differentiation networks and their regulation from time series single-cell expression data. Genome Res. 28, 38–395 (2018).
Chen, Y., Georgiou, T. T. & Pavon, M. The most likely evolution of diffusing and vanishing particles: Schrödinger bridges with unbalanced marginals. SIAM J. Control Optimiz. 60, 21M1447672 (2022).
Massri, A. J. et al. Developmental single-cell transcriptomics in the Lytechinus variegatus sea urchin embryo. Development 148, dev198614 (2021).
Weinreb, C., Rodriguez-Fraticelli, A., Camargo, F. D. & Klein, A. M. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science 367, eaaw3381 (2020).
Chen, W. et al. Live-seq enables temporal transcriptomic recording of single cells. Nature 608, 733–740 (2022).
Somnath, V. R. et al. Aligned diffusion Schrödinger bridges. In Proc. 39th Conf. Uncertainty in Artificial Intelligence Vol. 216, 1985–1995 (PMLR, 2023). Building on the connections of optimal transport to control theory, this method allows to reconstruct cellular dynamics that respects and integrates known trajectories, for example, obtained from DNA-barcoding technologies.
Kobayashi-Kirschvink, K. J. et al. Prediction of single-cell RNA expression profiles in live cells by Raman microscopy with Raman2RNA. Nat. Biotechnol. https://doi.org/10.1038/s41587-023-02082-2 (2024).
Vayer, T., Chapel, L., Flamary, R., Tavenard, R. & Courty, N. Fused Gromov–Wasserstein distance for structured objects. Algorithms 13, 212 (2020).
Zeira, R., Land, M., Strzalkowski, A. & Raphael, B. J. Alignment and integration of spatial transcriptomics data. Nat. Methods 19, 567–575 (2022). Using optimal transport methods for heterogeneous spaces known as the Gromov–Wasserstein problem, this paper allows the integration and alignment of spatial transcriptomics tissue slices.
Ma, Q. & Xu, D. Deep learning shapes single-cell data analysis. Nat. Rev. Mol. Cell Biol. 23, 303–304 (2022).
Ji, Y., Lotfollahi, M., Wolf, F. A. & Theis, F. J. Machine learning for perturbational single-cell omics. Cell Systems 12, 522–537 (2021).
Raimundo, F., Meng-Papaxanthos, L., Vallot, C. & Vert, J.-P. Machine learning for single-cell genomics data analysis. Curr. Opin. Syst. Biol. 26, 64–71 (2021).
Roohani, Y., Huang, K. & Leskovec, J. GEARS: predicting transcriptional outcomes of novel multi-gene perturbations. Nat. Biotechnol. 42, 927–935 (2023).
Amos, B. On amortizing convex conjugates for optimal transport. In Int. Conf. Learning Representations (ICLR, 2023).
Taghvaei, A. & Jalali, A. 2-Wasserstein approximation via restricted convex potentials with application to improved training for GANs. Preprint at https://arxiv.org/abs/1902.07197 (2019).
Lopez, R., Regier, J., Cole, M. B., Jordan, M. I. & Yosef, N. Deep generative modeling for single-cell transcriptomics. Nat. Methods 15, 1053–1058 (2018).
Lotfollahi, M., Wolf, F. A. & Theis, F. J. scGen predicts single-cell perturbation responses. Nat. Methods. 16, 715–721 (2019).
Lotfollahi, M. et al. Predicting cellular responses to complex perturbations in high-throughput screens. Mol. Syst. Biol. 19, e11517 (2023).
Waddington, C. H. The Strategy of the Genes: A Discussion of Some Aspects of Theoretical Biology (G. Allen and Unwin, 1957).
Schiebinger, G. Reconstructing developmental landscapes and trajectories from single-cell data. Curr. Opin. Syst. Biol. 27, 100351 (2021).
Lange, M. et al. Mapping lineage-traced cells across time points with moslin. Preprint at bioRxiv https://doi.org/10.1101/2023.04.14.536867 (2023). Building on the fused Gromov–Wasserstein distance, this paper introduces a method to reconstruct developmental processes based on both single-cell gene expressions and lineage information.
Prasad, N., Yang, K. & Uhler, C. Optimal transport using GANs for lineage tracing. Preprint at https://arxiv.org/abs/2007.12098 (2020).
Grathwohl, W., Chen, R. T., Bettencourt, J., Sutskever, I. & Duvenaud, D. FFJORD: free-form continuous dynamics for scalable reversible generative models. In Int. Conf. Learning Representations (ICLR, 2019).
Moon, K. R. et al. Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol. 37, 1482–1492 (2019).
Schrödinger, E. Über die Umkehrung der Naturgesetze (Verlag der Akademie der Wissenschaften in Kommission bei Walter De Gruyter u. Company, 1931).
Schrödinger, E. Sur la théorie relativiste de l’électron et l’interprétation de la mécanique quantique. Annales de l’institut Henri Poincaré 2, 269–310 (1932).
De Bortoli, V., Thornton, J., Heng, J. & Doucet, A. Diffusion Schrödinger bridge with applications to score-based generative modeling. In Advances in Neural Information Processing Systems Vol. 34 (NeurIPS, 2021).
Chen, T., Liu, G.-H. & Theodorou, E. A. Likelihood training of Schrödinger bridge using forward-backward SDEs theory. In Int. Conf. Learning Representations (ICLR, 2022).
Winkler, L., Ojeda, C. & Opper, M. A score-based approach for training Schrödinger bridges for data modelling. Entropy 25, 316 (2023).
Winkler, L., Ojeda, C. & Opper, M. 一种基于分数的训练薛定谔桥进行数据建模的方法。 熵 25 , 316 (2023)。Tong, A. et al. Improving and generalizing flow-based generative models with minibatch optimal transport. Trans. Mach. Learn. Res. (2024). By combining optimal transport and known concepts from optimal control, this paper presents a flow matching approach that allows to model cellular dynamics over time.
Tong, A. 等人。《利用小批量最优传输改进和推广基于流的生成模型》。 《机器学习与计算研究学报 》(2024 年)。 通过结合最优传输和最优控制中的已知概念,本文提出了一种流匹配方法,可以对细胞随时间变化的动态进行建模。Chen, T., Liu, G.-H., Tao, M. & Theodorou, E. A. Deep momentum multi-marginal Schrödinger bridge. In Advances in Neural Information Processing Systems (NeurIPS, 2023).
Chen, T., Liu, G.-H., Tao, M. & Theodorou, EA, 深度动量多边缘薛定谔桥。载于《神经信息处理系统进展》 (NeurIPS,2023)。Di Marino, S. & Chizat, L. A tumor growth model of Hele-Shaw type as a gradient flow. EESAIM Control Optim. Calc. Var. 26, 103 (2020).
Di Marino, S. & Chizat, L. Hele-Shaw 型梯度流肿瘤生长模型。EESAIM 控制优化计算变量。26 , 103 (2020)。Jiang, Q., Zhang, S. & Wan, L. Dynamic inference of cell developmental complex energy landscape from time series single-cell transcriptomic data. PLoS Comput. Biol. 18, e1009821 (2022).
Jiang, Q., Zhang, S. & Wan, L. 从时间序列单细胞转录组数据动态推断细胞发育复杂能量景观。PLoS Comput. Biol. 18 , e1009821 (2022)。Ambrosio, L., Gigli, N. & Savaré, G. Gradient Flows in Metric Spaces and in the Space of Probability Measures (Springer, 2006).
Ambrosio, L.、Gigli, N. 和 Savaré, G. 度量空间和概率测度空间中的梯度流 (Springer, 2006)。Risken, H. The Fokker–Planck Equation (Springer, 1996).
Risken, H. 福克-普朗克方程 (Springer,1996)。Jordan, R., Kinderlehrer, D. & Otto, F. The variational formulation of the Fokker–Planck equation. SIAM J. Math. Anal. 29, S0036141096303359 (1998).
Jordan, R., Kinderlehrer, D. 和 Otto, F. 福克-普朗克方程的变分公式。SIAM J. Math. Anal. 29 , S0036141096303359 (1998)。Benamou, J.-D., Carlier, G. & Laborde, M. An augmented Lagrangian approach to Wasserstein gradient flows and applications. ESAIM Proc. Surv. 54, hal-01245184 (2016).
Benamou, J.-D.、Carlier, G. 和 Laborde, M. 一种增强拉格朗日方法求解 Wasserstein 梯度流及其应用。ESAIM Proc. Surv. 54 , hal-01245184 (2016)。Carrillo, J. A., Craig, K., Wang, L. & Wei, C. Primal dual methods for Wasserstein gradient flows. Found. Computat. Math. 22, 389–443 (2021).
Carrillo, JA, Craig, K., Wang, L. & Wei, C. Wasserstein 梯度流的原始对偶方法 。《计算机数学》 22,389–443 (2021)。Peyré, G. Entropic approximation of Wasserstein gradient flows. SIAM J. Imaging Sci. 8, 15M1010087 (2015).
Peyré, G. Wasserstein 梯度流的熵近似。SIAM J. Imaging Sci. 8 , 15M1010087 (2015)。Otto, F. The geometry of dissipative evolution equations: the porous medium equation. Commun. Partial Differ. Equ. 26, 101–174 (2001).
Otto, F. 耗散演化方程的几何学:多孔介质方程。 《偏微分方程通报》 26 , 101–174 (2001)。Hashimoto, T., Gifford, D. & Jaakkola, T. Learning population-level diffusions with generative recurrent networks. In Int. Conf. Machine Learning Vol. 33 (ICML, 2016).
Hashimoto, T.、Gifford, D. 和 Jaakkola, T.,利用生成循环网络学习群体层面的扩散。刊于国际机器学习会议第 33 卷(ICML,2016)。Huguet, G. et al. Manifold interpolating optimal-transport flows for trajectory inference. In Advances in Neural Information Processing Systems Vol. 35 (NeurIPS, 2022).
Huguet, G. 等人。用于轨迹推断的流形插值最优传输流。载于《神经信息处理系统进展》 第 35 卷(NeurIPS,2022 年)。Lotfollahi, M., Naghipourfar, M., Theis, F. J. & Wolf, F. A. Conditional out-of-distribution generation for unpaired data using transfer VAE. Bioinformatics 36, i610–i617 (2020).
Lotfollahi, M.、Naghipourfar, M.、Theis, FJ 和 Wolf, FA,基于迁移变分自编码器 (VAE) 实现非配对数据的条件分布外生成。 《生物信息学》 36 ,i610–i617 (2020)。Torous, W., Gunsilius, F. & Rigollet, P. An optimal transport approach to causal inference. Preprint at https://arxiv.org/abs/2108.05858 (2021).
Torous, W.、Gunsilius, F. 和 Rigollet, P. 因果推理的最优传输方法。预印本网址: https://arxiv.org/abs/2108.05858 (2021)。Tu, R., Zhang, K., Kjellström, H. & Zhang, C. Optimal transport for causal discovery. In Int. Conf. Learning Representations (ICLR, 2022).
Tu, R.、Zhang, K.、Kjellström, H. 和 Zhang, C. 因果发现的最佳传输。发表于国际会议“学习表征” (ICLR,2022)。Abadie, A. Semiparametric difference-in-differences estimators. Rev. Econ. Stud. 72, 1–19 (2005).
Abadie, A. 半参数双重差分估计量。 《经济学研究评论》 72,1-19 (2005)。Singh, R. et al. Prioritizing transcription factor perturbations from single-cell transcriptomics. Preprint at bioRxiv https://doi.org/10.1101/2022.06.27.497786 (2022).
Singh, R. 等人 。从单细胞转录组学角度优先分析转录因子扰动。bioRxiv 预印本 https://doi.org/10.1101/2022.06.27.497786 ( 2022)。Yang, K. D. et al. Multi-domain translation between single-cell imaging and sequencing data using autoencoders. Nat. Commun. 12, 31 (2021).
Yang, KD 等人。使用自动编码器在单细胞成像和测序数据之间进行多域转换。 《自然通讯》 12 , 31 (2021)。Amodio, M. et al. Exploring single-cell data with deep multitasking neural networks. Nat. Methods 16, 1139–1145 (2019).
Amodio, M.等人。利用深度多任务神经网络探索单细胞数据。 《自然-方法》 16 , 1139–1145 (2019)。Luecken, M. D. et al. Benchmarking atlas-level data integration in single-cell genomics. Nat. Methods 19, 41–50 (2022).
Luecken, MD 等。单细胞基因组学中图谱级数据整合的基准测试。 《自然-方法》 19 , 41–50 (2022)。Cao, K., Gong, Q., Hong, Y. & Wan, L. A unified computational framework for single-cell data integration with optimal transport. Nat. Commun. 13, 7419 (2022).
Ryu, J., Lopez, R., Bunne, C. & Regev, A. Cross-modality matching and prediction of perturbation responses with labeled Gromov–Wasserstein optimal transport. Preprint at https://arxiv.org/abs/2405.00838 (2024).
Demetci, P., Santorella, R., Sandstede, B. & Singh, R. Unsupervised integration of single-cell multi-omics datasets with disproportionate cell-type representation. In Research in Computational Molecular Biology: 26th Annual International Conference, RECOMB (Springer, 2022).
Tran, Q. H. et al. Unbalanced CO-optimal transport. In AAAI Conf. Artificial Intelligence Vol. 37 (AAAI, 2023).
Ma, S. et al. Chromatin potential identified by shared single-cell profiling of RNA and chromatin. Cell 183, 1103–1116.e20 (2020).
Novershtern, N. et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell 144, 296–309 (2011).
Lara-Astiaso, D. et al. Chromatin state dynamics during blood formation. Science 345, 943–949 (2014).
Huizing, G.-J., Deutschmann, I. M., Peyre, G. & Cantini, L. Paired single-cell multi-omics data integration with Mowgli. Nat. Commun. 14, 7711 (2023).
Haghverdi, L., Lun, A. T., Morgan, M. D. & Marioni, J. C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat. Biotechnol. 36, 421–427 (2018).
Joodaki, M. et al. Detection of PatIent-Level distances from single cell genomics and pathomics data with optimal transport (PILOT). Mol. Syst. Biol. 20, 57–74 (2024).
Weinberger, E., Lopez, R., Huetter, J.-C. & Regev, A. Disentangling shared and group-specific variations in single-cell transcriptomics data with multiGroupVI. In Proc. 17th Machine Learning in Computational Biology Meeting Vol. 200 (PMLR, 2022).
Tong, A. Y. et al. Diffusion Earth mover’s distance and distribution embeddings. In Int. Conf. Machine Learning (ICML, 2021).
Tong, A. et al. Embedding signals on graphs with unbalanced diffusion Earth mover’s distance. In IEEE Int. Conf. Acoustics, Speech and Signal Processing (ICASSP) (IEEE, 2022).
Wang, Z. et al. QOT: efficient computation of sample level distance matrix from single-cell omics data through quantized optimal transport. Preprint at bioRxiv https://doi.org/10.1101/2024.02.06.578032 (2024).
Zapatero, M. R. et al. Trellis tree-based analysis reveals stromal regulation of patient-derived organoid drug responses. Cell 186, 5606–5619 (2023).
Rodriques, S. G. et al. Slide-seq: a scalable technology for measuring genome-wide expression at high spatial resolution. Science 363, 1463–1467 (2019).
Chen, K. H., Boettiger, A. N., Moffitt, J. R., Wang, S. & Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 348, aaa6090 (2015).
Shah, S., Lubeck, E., Zhou, W. & Cai, L. In situ transcription profiling of single cells reveals spatial organization of cells in the mouse hippocampus. Neuron 92, 342–357 (2016).
Rahimi, A., Vale-Silva, L. A., Savitski, M. F., Tanevski, J. & Saez-Rodriguez, J. DOT: a flexible multi-objective optimization framework for transferring features across single-cell and spatial omics. Nat. Commun. 15, 4994 (2024).
Alvarez-Melis, D., Jaakkola, T. & Jegelka, S. Structured optimal transport. In Int. Conf. Artificial Intelligence and Statistics (AISTATS, 2018).
Bradbury, J. et al. JAX: composable transformations of Python+NumPy programs. GitHub http://github.com/google/jax (2018).
Flamary, R. et al. POT: Python optimal transport. J. Mach. Learn. Res. 22, 1–8 (2021). A Python library providing both NumPy-based and PyTorch-based implementations of various optimal transport algorithms.
Harris, C. R. et al. Array programming with NumPy. Nature 585, 357–362 (2020).
Paszke, A. et al. PyTorch: an imperative style, high-performance deep learning library. In Advances in Neural Information Processing Systems Vol. 32 (NeurIPS, 2019).
Feydy, J. et al. Interpolating between optimal transport and MMD using Sinkhorn divergences. In Int. Conf. Artificial Intelligence and Statistics Vol. 22 (AISTATS, 2019).
Chen, A. et al. Spatiotemporal transcriptomic atlas of mouse organogenesis using DNA nanoball-patterned arrays. Cell 185, 1777–1792.e21 (2022).
Weinreb, C., Wolock, S., Tusi, B. K., Socolovsky, M. & Klein, A. M. Fundamental limits on dynamic inference from single-cell snapshots. Proc. Natl Acad. Sci. USA 115, E2467–E2476 (2018).
Pan, X., Li, H. & Zhang, X. TedSim: temporal dynamics simulation of single-cell RNA sequencing data and cell division history. Nucleic Acids Res. 50, 272–4288 (2022).
Packer, J. S. et al. A lineage-resolved molecular atlas of C. elegans embryogenesis at single-cell resolution. Science 365, eaax1971 (2019).
Hu, B. et al. Origin and function of activated fibroblast states during zebrafish heart regeneration. Nat. Genet. 54, 1227–1237 (2022).
Hagai, T. et al. Gene expression variability across cells and species shapes innate immunity. Nature 563, 197–202 (2018).
Zhao, W. et al. Deconvolution of cell type-specific drug responses in human tumor tissue with single-cell RNA-seq. Genome Med. 13, 82 (2021).
Kang, H. M. et al. Multiplexed droplet single-cell RNA-sequencing using natural genetic variation. Nat. Biotechnol. 36, 89–94 (2018).
Luecken, M. D. et al. A sandbox for prediction and integration of DNA, RNA, and proteins in single cells. In Proc. Neural Information Processing Systems Track on Datasets and Benchmarks 1 (Round 2) (NeurIPS, 2021).
Lance, C. et al. Multimodal single cell data integration challenge: results and lessons learned. Proceedings of the NeurIPS 2021 Competitions and Demonstrations Track Vol. 176, 162–176 (PMLR, 2022).
Codeluppi, S. et al. Spatial organization of the somatosensory cortex revealed by osmFISH. Nat. Methods 15, 932–935 (2018).
Liu, L. et al. Deconvolution of single-cell multi-omics layers reveals regulatory heterogeneity. Nat. Commun. 10, 470 (2019).
Luo, C. et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science 357, 600–604 (2017).
Regev, A. et al. The Human Cell Atlas. eLife 6, e27041 (2017).
Abdulla, S. et al. CZ CELL×GENE Discover: a single-cell data platform for scalable exploration, analysis and modeling of aggregated data. Preprint at bioRxiv https://doi.org/10.1101/2023.10.30.563174 (2023).
Megill, C. et al. CELL×GENE: a performant, scalable exploration platform for high dimensional sparse matrices. Preprint at bioRxiv https://doi.org/10.1101/2021.04.05.438318 (2021).
Peidli, S. et al. scPerturb: information resource for harmonized single-cell perturbation data. Nat. Methods 21, 531–540 (2024).
Sinkhorn, R. A relationship between arbitrary positive matrices and doubly stochastic matrices. Ann. Math. Statist. 35, 876–879 (1964).
Heydari, T. et al. IQCELL: a platform for predicting the effect of gene perturbations on developmental trajectories using single-cell RNA-seq data. PLoS Comput. Biol. 18, e1009907 (2022).
Busch, K. et al. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 518, 542–546 (2015).
Xiong, Y.-X. & Zhang, X.-F. scdot: enhancing single-cell RNA-seq data annotation and uncovering novel cell types through multi-reference integration. Brief. Bioinform. 25, bbae072 (2024).
Lipman, Y., Chen, R. T., Ben-Hamu, H., Nickel, M. & Le, M. Flow matching for generative modeling. In Int. Conf. Learning Representations (ICLR, 2023).
Liu, X., Wu, L., Ye, M. & Liu, Q. Flow straight and fast: learning to generate and transfer data with rectified flow. In International Conference on Learning Representations (ICLR) (2023).
Pooladian, A.-A. et al. Multisample flow matching: straightening flows with minibatch couplings. In Int. Conf. Machine Learning (ICML, 2023).
Albergo, M. S., Boffi, N. M. & Vanden-Eijnden, E. Stochastic interpolants: a unifying framework for flows and diffusions. Preprint at https://arxiv.org/abs/2303.08797 (2023).
Liu, G.-H. et al. I2 SB: image-to-image Schrödinger bridge. In Int. Conf. Machine Learning (ICML, 2023).
Liu, G.-H., Chen, T., So, O. & Theodorou, E. A. Deep generalized Schrödinger bridge. In Advances in Neural Information Processing Systems (NeurIPS, 2022).
Brandstetter, J., Worrall, D. & Welling, M. Message passing neural PDE solvers. In Int. Conf. Learning Representations (ICLR, 2022).
Raissi, M., Perdikaris, P. & Karniadakis, G. E. Physics-informed neural networks: a deep learning framework for solving forward and inverse problems involving nonlinear partial differential equations. J. Comput. Phys. 378, 686–707 (2019).
Song, Y. et al. Score-based generative modeling through stochastic differential equations. In Int. Conf. Learning Representations (ICLR, 2021).
Daniels, M., Maunu, T. & Hand, P. Score-based generative neural networks for large-scale optimal transport. In Advances in Neural Information Processing Systems Vol. 34 (NeurIPS, 2021).
Kong, Z., Ping, W., Huang, J., Zhao, K. & Catanzaro, B. DiffWave: a versatile diffusion model for audio synthesis. In Int. Conf. Learning Representations (ICLR, 2021).
Song, Y. & Ermon, S. Generative modeling by estimating gradients of the data distribution. In Advances in Neural Information Processing Systems (NeurIPS, 2019).
Comiter, C. et al. Inference of single cell profiles from histology stains with the single-cell omics from histology analysis framework (SCHAF). Preprint at bioRxiv https://doi.org/10.1101/2023.03.21.533680 (2023).
Rosen, Y. et al. Universal cell embeddings: a foundation model for cell biology. Preprint at bioRxiv https://doi.org/10.1101/2023.11.28.568918 (2023).
Cui, H. et al. scGPT: toward building a foundation model for single-cell multi-omics using generative AI. Nat. Methods https://doi.org/10.1038/s41592-024-02201-0 (2024).
Chan, E. M. et al. Live cell imaging distinguishes bona fide human iPS cells from partially reprogrammed cells. Nat. Biotechnol. 27, 1033–1037 (2009).
Shi, Y., De Bortoli, V., Campbell, A. & Doucet, A. Diffusion Schrödinger bridge matching. In Advances in Neural Information Processing Systems (NeurIPS, 2024).
Irmisch, A. et al. The Tumor Profiler Study: integrated, multi-omic, functional tumor profiling for clinical decision support. Cancer Cell 39, 288–293 (2021).
Santinha, A. J. et al. Transcriptional linkage analysis with in vivo AAV-Perturb-seq. Nature 622, 367–375 (2023).
Cleary, B., Cong, L., Cheung, A., Lander, E. S. & Regev, A. Efficient generation of transcriptomic profiles by random composite measurements. Cell 171, 1424–1436.e18 (2017).
Cleary, B. & Regev, A. The necessity and power of random, under-sampled experiments in biology. Preprint at https://arxiv.org/abs/2012.12961 (2020).
Frangieh, C. J. et al. Multimodal pooled Perturb-CITE-seq screens in patient models define mechanisms of cancer immune evasion. Nat. Genet. 53, 332–341 (2021).
Wu, F. et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat. Commun. 12, 2540 (2021).
González-Silva, L., Quevedo, L. & Varela, I. Tumor functional heterogeneity unraveled by scRNA-seq technologies. Trends Cancer 6, 13–19 (2020).
Li, C. et al. Single-cell transcriptomics reveals cellular heterogeneity and molecular stratification of cervical cancer. Commun. Biol. 5, 1208 (2022).
Bertsimas, D. & Tsitsiklis, J. Introduction to Linear Optimization (Athena Scientific, 1997).
Franklin, J. & Lorenz, J. On the scaling of multidimensional matrices. Linear Algebra Appl. 114, 717–735 (1989).
Acknowledgements
This publication was supported by the NCCR Catalysis (grant number 180544), a National Centre of Competence in Research funded by the Swiss National Science Foundation.
Ethics declarations
Competing interests
C.B. and A.R. are employees of Genentech. A.R. is a co-founder and equity holder of Celsius Therapeutics, an equity holder in Immunitas Therapeutics and, until 31 July 2020, was a scientific advisory board member of Thermo Fisher Scientific, Syros Pharmaceuticals, Asimov and Neogene Therapeutics. A.R. is a named inventor on multiple patents related to single-cell and spatial genomics, including AI methods applied to such data.
Peer review
Peer review information
Nature Reviews Methods Primers thanks Ivan Costa, Liang Ma, James Nagai, Lin Wan, Daifeng Wang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Related links
GeomLoss: https://www.kernel-operations.io/geomloss/
Multiomics Single-cell Optimal Transport (moscOT): https://moscot.readthedocs.io/
Optimal Transport Toolbox (OTT): https://ott-jax.readthedocs.io/
Python Optimal Transport (POT): https://pythonot.github.io/
Glossary
- Autoencoder
-
Deep neural network consisting of an encoder and decoder network that learns efficient codings of unlabelled data in an unsupervised fashion.
- Bijective
-
A function that is both injective (one-to-one) and surjective (onto), ensuring a perfect pairing between elements of the source and target domain.
- Confounding factor
-
A variable in causal inference that influences both the dependent variable and the independent variable, causing a spurious association, masking an actual association, or falsely demonstrating an apparent association between the study variables.
- Constrained optimization problem
-
An optimization problem that includes constraints on the variables, often taking the form of equalities or inequalities that restrict the feasible region.
- Coupling
-
A coupling of μ and ν is a probability measure on the product space of their respective supports, such that its first and second marginals coincide with μ and ν.
- Dualize
-
The process of converting a constrained optimization problem into an unconstrained one, carried out by transforming constraints into penalty terms in the objective function.
- Flows
-
The continuous transformation of one distribution into another.
- Geodesic flow
-
The smooth path connecting two points in a given space, often representing the shortest distance between them.
- Matching
-
The process of pairing bijectively elements from two families of points of the same size, using a permutation.
- Normalizing flows
-
A deep learning architecture that constructs complex distributions by transforming a probability density through a series of invertible mappings.
- Optimal transport
-
A mathematical theory describing tools to infer associations, alignments or mappings between unaligned point clouds μ and ν, for example, two measurement snapshots or data sets derived from single-cell or spatial omics technologies.
- Pushforward map
-
Given a vector-to-vector function F, the pushforward map associates to any measure μ the pushforward measure T♯μ. The pushforward map, therefore, lifts a regular vector-to-vector function F into an operator that takes a probability distribution to output another distribution.
- Transport map
-
A vector-to-vector function T that associates to each point x in the support of a source distribution another point T(x) that is in the support of a target distribution, while satisfying a pushforward constraint T♯μ = ν.
- Transport plan
-
A coupling (either in matrix or joint density form), quantifying the strength of association between any point x in the source distribution μ and target point y in the ν distribution, as P(x, y).
- Twist condition
-
Given a cost function c(x, y) taking two input vectors, this refers to the requirement that at any given point x, the map y → ∇1c(x, y) be invertible. Although not necessary, this condition simplifies many proofs when proving the existence of optimal transport map.
- Unbalanced association
-
A generalization of coupling defined to bring more flexibility to optimal transport computations. Such a generalization arises when considering unnormalized probability distributions on the product space of the supports μ and ν, without requiring that its marginal coincides exactly with μ and ν.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bunne, C., Schiebinger, G., Krause, A. et al. Optimal transport for single-cell and spatial omics. Nat Rev Methods Primers 4, 58 (2024). https://doi.org/10.1038/s43586-024-00334-2
Accepted:
Published:
DOI: https://doi.org/10.1038/s43586-024-00334-2