

Abstract 摘要
Immune microenvironment could affect the biological progress in prostate cancer (PCa) through N6 methyl adenosine (m6A) methylation. The purpose of this study was to investigate the crosstalk between m6A methylation and immune microenvironment and explore potential biomarkers to improve the immunotherapeutic response. Firstly, according to 11 differentially expressed m6A genes between normal and tumor samples, PCa patients were divided into immune microenvironment subtype 1 (IMS1) and IMS2 based on m6A gene profiles extracted from The Cancer Genome Atlas (TCGA) database. IMS2 showed an immune “cold” phenotype with worse prognoses, and HNRNPC was identified as the biomarker of IMS2 by the protein‐protein interaction network. Furthermore, through bioinformatics analyses and in vitro experiments, we found that HNRNPC‐high patients showed a suppressive immune‐infiltrating tumor microenvironment with a higher infiltration of regulatory T (Treg) cells. Finally, we cocultured transfected PCa cells with peripheral blood mononuclear cells (PBMC) and verified that HNRNPC inhibits tumor immunity by elevating the activation of Treg cells and suppression of effector CD8 T cell. In conclusion, we identified a “cold” immune phenotype in PCa, and HNRNPC regulating the activation of Treg cells. Activation of the immune microenvironment through targeting HNRNPC may be a potential therapeutic option for advanced PCa.
免疫微环境可能通过 N6 甲基腺苷(m6A)甲基化影响前列腺癌(PCa)的生物进程。本研究的目的是探讨 m6A 甲基化与免疫微环境之间的相互作用,并探索潜在的生物标志物以改善免疫治疗反应。首先,根据正常样本和肿瘤样本之间 11 个差异表达的 m6A 基因,PCa 患者根据从癌症基因组图谱(TCGA)数据库提取的 m6A 基因谱被分为免疫微环境亚型 1(IMS1)和 IMS2。IMS2 表现出免疫“冷”表型,预后较差,HNRNPC被识别为 IMS2 的生物标志物,通过蛋白质-蛋白质相互作用网络进一步确认。此外,通过生物信息学分析和体外实验,我们发现HNRNPC高表达的患者表现出抑制性免疫浸润肿瘤微环境,调节性 T 细胞(Treg 细胞)的浸润水平更高。 最后,我们将转染的前列腺癌细胞与外周血单核细胞(PBMC)共同培养,并验证了HNRNPC通过提高 Treg 细胞的活化和抑制效应 CD8 T 细胞来抑制肿瘤免疫。总之,我们在前列腺癌中识别出了一种“冷”免疫表型,并且HNRNPC调节 Treg 细胞的活化。通过靶向HNRNPC激活免疫微环境可能是晚期前列腺癌的一种潜在治疗选择。
Keywords: HNRNPC, M6A methylation, prostate cancer, regulatory T (Treg) cells, tumor immune microenvironment
关键词:HNRNPC,M6A 甲基化,前列腺癌,调节性 T 细胞(Treg),肿瘤免疫微环境
Harboring a high expression level of m6A reader HNRNPC, progressed prostate cancer (PCa) exhibited a “cold” immune phenotype, which was mainly induced by increasing Treg abundance. Targeting HNRNPC may help activate the immune microenvironment or elevate the sensitiveness to anti‐CTLA4 therapies.
携带高表达水平的 m6A 读取器 HNRNPC 的前列腺癌(PCa)表现出“冷”免疫表型,这主要是由于 Treg 丰度的增加所致。靶向 HNRNPC 可能有助于激活免疫微环境或提高对抗 CTLA4 治疗的敏感性。
Abbreviations 缩写
- AS
aneuploidy score 非整倍体评分
- BCR
B cell receptor B 细胞受体
- BCR
biochemical recurrence 生化复发
- BH
Hochberg 霍赫伯格
- CTLA‐4
cytotoxic T lymphocyte associated protein 4
细胞毒性 T 淋巴细胞相关蛋白 4- DEMG
differentially expressed m6A gene
差异表达的 m6A 基因- FDR
false discovery rate 假阳性率
- GEO 地理
Gene Expression Omnibus 基因表达综合数据库
- HLA
human leukocyte antigen 人类白细胞抗原
- hnRNA
heteronuclear RNA 异核 RNA
- hnRNP
heterogeneous nuclear ribonucleoprotein
异质核核糖核蛋白- HRD 人力资源发展
homologous recombination defects
同源重组缺陷- ICD 国际疾病分类
immunogenic cell death 免疫原性细胞死亡
- ICI
immune checkpoint inhibitor
免疫检查点抑制剂- ICP
immune checkpoints 免疫检查点
- IMS
immune microenvironment subtype
免疫微环境亚型- IRG
immune‐related genes 免疫相关基因
- ITH
intratumor heterogeneity 肿瘤内异质性
- iTreg
induced regulatory T cells
诱导性调节性 T 细胞- m6A
N6 methyl adenosine N6 甲基腺苷
- mCRPC
metastatic castration‐resistant prostate cancer
转移性去势抵抗性前列腺癌- MDSC
myeloid‐derived suppressor cell
髓源抑制细胞- MHC
major histocompatibility complex
主要组织相容性复合体- NK
natural killer cell 自然杀伤细胞
- NKT
natural killer T cell 自然杀伤 T 细胞
- nTreg
natural regulatory T cell
自然调节性 T 细胞- PCa 前列腺癌
prostate cancer 前列腺癌
- PD‐1
programmed death‐1 receptor
程序性死亡-1 受体- PD‐L1
programmed death‐1 ligand
程序性死亡-1 配体- PPI 生产者物价指数
protein‐protein interaction
蛋白质-蛋白质相互作用- PRAD
prostate adenocarcinoma 前列腺腺癌
- TCGA
The Cancer Genome Atlas 癌症基因组图谱
- TCIA
The Cancer Immunome Atlas
癌症免疫组图谱- TCR
T cell receptor T 细胞受体
- Th1
T helper 1 cell T 辅助 1 细胞
- TMB
tumor mutation burden 肿瘤突变负担
- TME
tumor microenvironment 肿瘤微环境
- TPM
transcripts per million 每百万转录本
- UTR
untranslated region 未翻译区域
- WES
whole‐exome sequencing 全外显子测序
1. INTRODUCTION 1. 引言
Prostate cancer (PCa) is a common malignancy in men.
1
,
2
Despite the rapid response to androgen deprivation therapy, most PCa patients eventually progress to fatal metastatic castration‐resistant PCa (mCRPC).
3
,
4
Recently, immune checkpoint inhibitors (ICIs) have become choices for progressive PCa.
5
,
6
Although immunotherapy has proven to be an effective and important new strategy for the management of PCa patients, only a few patients benefit from immunotherapy.
7
,
8
,
9
,
10
This phenomenon may be attributed to the varied heterogeneity of the immune microenvironment among individuals.
11
,
12
Therefore, it is important to further explore the regulatory mechanisms of the tumor immune microenvironment to optimize the management of immunotherapy.
前列腺癌(PCa)是男性常见的恶性肿瘤。 1 , 2 尽管对雄激素剥夺治疗反应迅速,但大多数 PCa 患者最终会进展为致命的转移性去势抵抗性前列腺癌(mCRPC)。 3 , 4 最近,免疫检查点抑制剂(ICIs)已成为进展性 PCa 的治疗选择。 5 , 6 尽管免疫治疗已被证明是管理 PCa 患者的有效且重要的新策略,但只有少数患者从免疫治疗中受益。 7 , 8 , 9 , 10 这种现象可能归因于个体之间免疫微环境的异质性差异。 11 , 12 因此,进一步探索肿瘤免疫微环境的调控机制以优化免疫治疗的管理非常重要。
N6‐methyladenosine (m6A) is the most common post‐transcriptional modification of mRNA and mediates more than 60% of RNA methylation.
13
,
14
The abnormal methylation level of m6A is closely related to stem cell differentiation and the immune response, which plays an important role in the progression of various cancers.
15
,
16
,
17
,
18
The abundance of m6A methylation modification in tumors mainly depends on the expression of methylation regulators, including methyltransferases (“writers”) and demethylases (“erasers”) in cells, while binding proteins (“readers”) perform a series of biological functions by binding to the methylation sites of m6A.
19
N6-甲基腺苷(m6A)是 mRNA 中最常见的转录后修饰,介导超过 60%的 RNA 甲基化。 13 , 14 m6A 的异常甲基化水平与干细胞分化和免疫反应密切相关,在各种癌症的进展中发挥着重要作用。 15 , 16 , 17 , 18 肿瘤中 m6A 甲基化修饰的丰度主要依赖于甲基化调节因子的表达,包括细胞中的甲基转移酶(“写入者”)和去甲基化酶(“擦除者”),而结合蛋白(“读取者”)通过与 m6A 的甲基化位点结合执行一系列生物功能。 19
In a previous study, Thorsson et al identified six “immune subtypes”: wound‐healing, IFN‐γ–dominant, inflammatory, lymphocyte‐depleted, immunologically quiet, and TGF‐β–dominant, which play different characters of immune features.
20
Currently, several methods have emerged to characterize the immune tumor microenvironment (TME) for the assessment of total lymphocytic infiltrates, including immune gene expression signatures, such as immunogenic cell death (ICD) and immune checkpoint (ICP) genes, neoantigen prediction, T cell receptor (TCR) and B cell receptor (BCR) repertoire inference, and somatic DNA alterations.
21
,
22
,
23
These methods and strategies provide a new basis for our research into the immune microenvironment of PCa.
在之前的一项研究中,Thorsson 等人识别了六种“免疫亚型”:伤口愈合型、IFN-γ主导型、炎症型、淋巴细胞耗竭型、免疫学安静型和 TGF-β主导型,这些亚型在免疫特征上扮演着不同的角色。 20 目前,已经出现了几种方法来表征免疫肿瘤微环境(TME),以评估总淋巴细胞浸润,包括免疫基因表达特征,如免疫原性细胞死亡(ICD)和免疫检查点(ICP)基因、新抗原预测、T 细胞受体(TCR)和 B 细胞受体(BCR)库推断,以及体细胞 DNA 改变。 21 , 22 , 23 这些方法和策略为我们研究前列腺癌的免疫微环境提供了新的基础。
In this study, we systematically evaluated the expression profile of m6A methylation regulators in PCa to improve the risk stratification of prognosis and promote therapeutic decision‐making in PCa. The relationship between m6A regulatory factors and immune cell infiltration was analyzed in silico and verified in vitro. Our study attempted to illustrate the m6A methylation regulatory mechanism and the prostate tumor immune microenvironment status of PCa, thus providing a basis for improving immunotherapy strategies in PCa.
在本研究中,我们系统地评估了前列腺癌中 m6A 甲基化调节因子的表达谱,以改善预后风险分层并促进前列腺癌的治疗决策。我们通过计算分析了 m6A 调节因子与免疫细胞浸润之间的关系,并在体外进行了验证。我们的研究试图阐明 m6A 甲基化调节机制及前列腺癌的肿瘤免疫微环境状态,从而为改善前列腺癌的免疫治疗策略提供基础。
2. MATERIALS AND METHODS 2. 材料与方法
2.1. Data processing 2.1. 数据处理
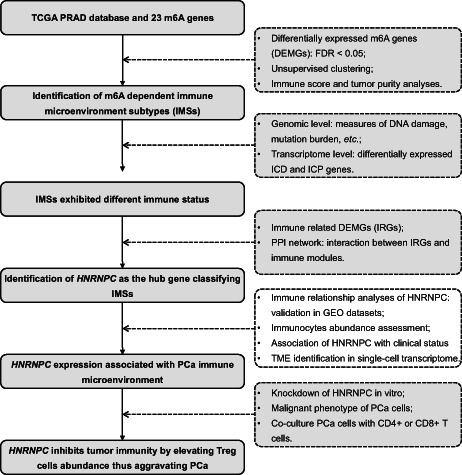
Figure 1 shows a flowchart of this study. The mRNA expression profiles and related clinical data were collected from The Cancer Genome Atlas (TCGA) data portal and Gene Expression Omnibus (GEO) datasets (GSE70768, GSE32571, GSE60329, GSE46602). The mRNA expression data were transformed to values in transcripts per million (TPM). A total of 23 m6A genes were integrated from several studies, as shown in Figure S1. The single‐cell transcriptome data were extracted from GEO dataset: GSE141445 including 13 samples, 12 primary samples, and 1 metastatic sample.
图 1 显示了本研究的流程图。mRNA 表达谱和相关临床数据来自癌症基因组图谱 (TCGA) 数据门户和基因表达综合数据库 (GEO) 数据集 (GSE70768, GSE32571, GSE60329, GSE46602)。mRNA 表达数据被转换为每百万转录本 (TPM) 的值。总共整合了 23 个 m6A 基因,来自多个研究,如图 S1 所示。单细胞转录组数据来自 GEO 数据集:GSE141445,包括 13 个样本,12 个原发样本和 1 个转移样本。
FIGURE 1. 图 1。
Flow chart of this study
本研究的流程图
2.2. Differentially expressed m6A genes (DEMGs) and cluster analysis
2.2. 差异表达的 m6A 基因 (DEMGs) 和聚类分析
We used Wilcoxon rank sum test to perform the differential expression analyses. M6A genes that met the criterion of false discovery rate (FDR) <0.05 between tumor and normal samples were identified as DEMGs. Based on the expression level of DEMGs, patients were then separated into subgroups using unsupervised clustering (named immune microenvironment subtypes [IMSs] subsequently).
我们使用 Wilcoxon 秩和检验进行差异表达分析。满足肿瘤和正常样本之间假发现率(FDR)<0.05 标准的 M6A 基因被确定为差异表达基因(DEMGs)。根据 DEMGs 的表达水平,患者随后通过无监督聚类分为亚组(随后称为免疫微环境亚型[IMSs])。
Differences in the clinical characteristics of patients between IMSs were measured, including clinical M stage, pathological T stage, pathological N stage, Gleason score, and biochemical recurrence (BCR). In addition, we investigated the differences in tumor purity and immune scores between clusters. Next, we calculated the ssGSEA score of immunostimulatory and inhibitory states using gene lists collected from previous studies, which are provided in Table S1.
在 IMS 之间测量患者的临床特征差异,包括临床 M 期、病理 T 期、病理 N 期、Gleason 评分和生化复发(BCR)。此外,我们还研究了不同簇之间肿瘤纯度和免疫评分的差异。接下来,我们使用从之前研究中收集的基因列表计算了免疫刺激和抑制状态的 ssGSEA 评分,这些基因列表在表S1中提供。
2.3. Identifying the immune landscape of IMS clusters
2.3. 识别 IMS 簇的免疫环境
First, we counted the proportion of C1‐C6 immune subtypes in the two IMSs. Considering that immune infiltration was related to “measures of DNA damage,” we compared several measures of DNA damage, including fraction alteration, number of segments, aneuploidy score (AS), homologous recombination defects (HRD), and intratumor heterogeneity (ITH) between IMSs. Next, a comparison was performed of the genome changes, including silent/nonsilent mutation, indel neoantigens, and SNV neoantigen counts. Differences in BCR and TCR diversity between clusters were also measured using the Shannon index. All data used were obtained from Thorsson et al
20
Finally, we compared the expression levels of ICD and ICP genes between the two IMSs.
首先,我们计算了两个 IMS 中 C1-C6 免疫亚型的比例。考虑到免疫浸润与“DNA 损伤的测量”相关,我们比较了几种 DNA 损伤的测量,包括分数改变、片段数量、非整倍体评分(AS)、同源重组缺陷(HRD)和肿瘤内异质性(ITH)。接下来,我们对基因组变化进行了比较,包括沉默/非沉默突变、插入缺失新抗原和 SNV 新抗原计数。还使用香农指数测量了不同簇之间 BCR 和 TCR 多样性的差异。所有使用的数据均来自 Thorsson 等人 20 。最后,我们比较了两个 IMS 之间 ICD 和 ICP 基因的表达水平。
Furthermore, whole‐exome sequencing (WES) from TCGA database data was used to calculate the tumor mutation burden (TMB) score in PCa with exons uniformly counted as 40 M regions and to describe the mutation spectrum of two clusters to search for specific mutations.
此外,使用 TCGA 数据库的数据进行全外显子测序(WES)来计算前列腺癌(PCa)的肿瘤突变负荷(TMB)评分,外显子统一计为 40 M 区域,并描述两个簇的突变谱以寻找特定突变。
2.4. Correlations of DEMGs with immune microenvironment
2.4. DEMGs 与免疫微环境的相关性
We performed correlation analyses of DEMGs with immune score and tumor purity to identify genes related to the immune microenvironment (named immune‐related genes [IRGs]). Genes with expression levels negatively correlated with immune score and positively correlated with tumor purity were determined as IRGs. To assess the outcome of immunotherapy for IMSs, we compared the expression of the following marker genes, which have been proven to affect immunotherapy in different ways: CD274 (PD‐L1),
21
CXCL9,
24
MEX3B,
25
HAVCR2 (TIM3),
22
CTLA4, and CD38.
26
我们对与免疫评分和肿瘤纯度相关的差异表达免疫相关基因(DEMGs)进行了相关性分析,以识别与免疫微环境相关的基因(称为免疫相关基因 [IRGs])。那些表达水平与免疫评分呈负相关而与肿瘤纯度呈正相关的基因被确定为 IRGs。为了评估免疫治疗对 IMS 的结果,我们比较了以下标记基因的表达,这些基因已被证明以不同方式影响免疫治疗:CD274 (PD‐L1), 21 CXCL9, 24 MEX3B, 25 HAVCR2 (TIM3), 22 CTLA4,和 CD38. 26
2.5. Identification of hub genes
2.5. 中心基因的识别
The STRING database (https://string‐db.org/) and Cytoscape software were used to establish a protein‐protein interaction (PPI) network with a minimum required interaction score of 0.40 to evaluate the interactions between proteins coded by IRGs and immune modules, including the major histocompatibility complex (MHC) module, receptor module, chemokine module, immunostimulatory module, and immunoinhibitory module. IRGs that interacted with immune modules were identified as hub genes.
STRING 数据库 (https://string‐db.org/) 和 Cytoscape 软件被用来建立一个蛋白质-蛋白质相互作用 (PPI) 网络,最低所需相互作用评分为 0.40,以评估 IRGs 编码的蛋白质与免疫模块之间的相互作用,包括主要组织相容性复合体 (MHC) 模块、受体模块、趋化因子模块、免疫刺激模块和免疫抑制模块。与免疫模块相互作用的 IRGs 被确定为中心基因。
2.6. Validation of hub genes through in silico analyses
2.6. 通过计算机分析验证中心基因
First, GEO datasets (GSE70768, GSE32571, GSE60329, and GSE46602) were applied to validate the correlations between IRGs and the immune microenvironment. Second, we analyzed the correlation between the expression of hub genes and MEX3B, which can downregulate HLA‐A expression on the surface of tumor cells, thereby rendering the tumor cells unable to be recognized and killed by T cells.
25
Next, tumor patients were separated into a high‐expression group and a low‐expression group according to the median hub gene expression levels; differences in the abundance of immune cells between the two groups were measured. Finally, based on TCGA database, we compared the differences in the expression levels of hub genes between different clinical conditions. Kaplan‐Meier (K‐M) plots were developed to perform survival analysis of hub genes.
首先,GEO 数据集(GSE70768,GSE32571,GSE60329 和 GSE46602)被应用于验证 IRGs 与免疫微环境之间的相关性。其次,我们分析了中心基因的表达与 MEX3B 之间的相关性,MEX3B 可以下调肿瘤细胞表面 HLA‐A 的表达,从而使肿瘤细胞无法被 T 细胞识别和杀死。 25 接下来,肿瘤患者根据中心基因表达水平的中位数被分为高表达组和低表达组;测量了两组之间免疫细胞丰度的差异。最后,基于 TCGA 数据库,我们比较了不同临床条件下中心基因表达水平的差异。Kaplan‐Meier (K‐M) 图被开发用于对中心基因进行生存分析。
2.7. Single‐cell transcriptome data analyses
2.7. 单细胞转录组数据分析
The single‐cell transcriptome data were preprocessed using the “Seurat” package. Well‐established markers for each cell type of PCa, integrated with the automatic cell annotation tool “SingleR,”
27
were applied to annotate the cell types, dividing the cells into luminal epithelial cells (AR), basal epithelial cells (TP63), T cells (CD3E), B cells (CD19), fibroblasts (MYL9), mast cells (KIT), monocytes (CD14), and endothelial cells (CD34). We calculated percentages of cell types among samples and analyzed their correlation with HNRNPC‐positive cell rate. “ProjecTIL” was used to parse human scRNA‐seq T cell data in the context of murine TIL profiles.
28
We then compared the difference of T cell subgroup composition between HNRNPC‐positive and ‐negative cells.
单细胞转录组数据使用“Seurat”包进行了预处理。针对前列腺癌(PCa)每种细胞类型的成熟标记,与自动细胞注释工具“SingleR,” 27 结合,应用于细胞类型的注释,将细胞分为腔道上皮细胞(AR)、基底上皮细胞(TP63)、T 细胞(CD3E)、B 细胞(CD19)、成纤维细胞(MYL9)、肥大细胞(KIT)、单核细胞(CD14)和内皮细胞(CD34)。我们计算了样本中细胞类型的百分比,并分析了它们与HNRNPC‐阳性细胞比例的相关性。“ProjecTIL”用于解析人类 scRNA-seq T 细胞数据,以小鼠 TIL 特征为背景。 28 然后,我们比较了HNRNPC‐阳性和阴性细胞之间 T 细胞亚群组成的差异。
We used “CellChat” R package to study cell‐cell communication.
29
The samples were divided into positive and negative groups according to whether the positive rate of HNRNPC was greater than 70%. We compared the strength of interaction among different cell types.
我们使用“CellChat” R 包来研究细胞间通信。 29 根据 HNRNPC 的阳性率是否大于 70%,将样本分为阳性组和阴性组。我们比较了不同细胞类型之间的相互作用强度。
2.8. Cell lines and cell culture
2.8. 细胞系和细胞培养
Prostate cancer cells (C4‐2B, LNCaP, DU145, and PC3) and normal human prostate epithelial cells (RWPE‐1) were purchased from the American Type Culture Collection (ATCC). All cells were tested and confirmed to be free of mycoplasma contamination before use. C4‐2B cells were cultured in DMEM/F12 (4:1). LNCaP cells were cultured in RPMI‐1640. DU145 cells were cultured in MEM. PC3 cells were cultured in the F‐12 K medium. RWPE‐1 cells were cultured in keratinocyte serum‐free medium (K‐SFM). All media were provided by Gibco and were treated with 10% FBS and 100 U/mL penicillin/streptomycin in a 5% CO2 incubator.
前列腺癌细胞(C4‐2B、LNCaP、DU145 和 PC3)和正常人前列腺上皮细胞(RWPE‐1)均购自美国典型培养物保藏中心(ATCC)。所有细胞在使用前均经过检测,确认无支原体污染。C4‐2B 细胞在 DMEM/F12(4:1)中培养。LNCaP 细胞在 RPMI‐1640 中培养。DU145 细胞在 MEM 中培养。PC3 细胞在 F‐12 K 培养基中培养。RWPE‐1 细胞在无角质形成细胞血清培养基(K‐SFM)中培养。所有培养基均由 Gibco 提供,并在 5% CO2 培养箱中处理了 10% FBS 和 100 U/mL 青霉素/链霉素。
2.9. Cell transfection 2.9. 细胞转染
Three specific shRNAs targeting HNRNPC and scrambled shRNA were synthesized by GenePharma. Cells were seeded in six‐well plates at a density of 8 × 106 cells/well, and the plates were placed at 37°C with 5% CO2. When cell confluence reached 70%, transfections were performed using the Lipofectamine 3000 kit (Invitrogen) according to the manufacturer's instructions.
三种特定的 shRNA 靶向HNRNPC,以及随机 shRNA 由 GenePharma 合成。细胞以 8 × 106个细胞/孔的密度接种在六孔板中,板子放置在 37°C、5% CO2的环境中。当细胞融合度达到 70%时,按照制造商的说明使用 Lipofectamine 3000 试剂盒(Invitrogen)进行转染。
2.10. Quantitative real‐time PCR (RT‐qPCR)
2.10. 定量实时 PCR (RT-qPCR)
Cells were seeded in six‐well plates at a density of 1.5 × 106 cells/well and incubated for 24 hours. Total RNA was extracted from the cells using TRIzol® reagent (Invitrogen), and the extracted RNA was reverse‐transcribed into cDNA using the ReverTra Ace qPCR RT Kit (Toyobo). Quantitative PCR was performed using SYBR® qPCR Mix (Toyobo) based on the 2−ΔΔCt method. GAPDH and U6 were used as the endogenous control genes. All experiments were performed in triplicates.
细胞以每孔 1.5 × 106个细胞的密度接种在六孔板中,并孵育 24 小时。使用 TRIzol®试剂(Invitrogen)从细胞中提取总 RNA,提取的 RNA 使用 ReverTra Ace qPCR RT Kit(Toyobo)反转录为 cDNA。基于 2−ΔΔCt方法,使用 SYBR® qPCR Mix(Toyobo)进行定量 PCR。GAPDH 和 U6 被用作内源性对照基因。所有实验均以三次重复进行。
2.11. Cell‐counting kit‐8 (CCK‐8) assays
2.11. 细胞计数试剂盒-8 (CCK-8) 检测
Cell viability was determined using a CCK‐8 kit (Beyotime Institute of Biotechnology), following the manufacturer's instructions. The cells (2 × 104 cells/well) were seeded in a 96‐well plate. After cell growth for 12, 24, 48, or 72 hours, 10 μL of CCK‐8 solution was added to each well. Two hours later, absorbance was measured at 450 nm using a microplate reader. All experiments were performed in triplicates.
细胞活力使用 CCK-8 试剂盒(碧云天生物科技)进行测定,按照制造商的说明进行操作。将细胞(2 × 104 细胞/孔)接种在 96 孔板中。经过 12、24、48 或 72 小时的细胞生长后,向每个孔中添加 10 μL CCK-8 溶液。两小时后,使用微孔板读数仪在 450 nm 处测量吸光度。所有实验均以三次重复进行。
2.12. EdU assays 2.12. EdU 检测
The cells were cultured in 24‐well plates until 70% confluence. After 10 μL EdU solution was added and 2 hours incubation, the cells were fixed with 4% paraformaldehyde. After washing, a Click‐iTR EdU kit was used to detect EdU. The nuclei were stained with DAPI and the cells were observed using a fluorescence microscope (Olympus). All experiments were performed in triplicates.
细胞在 24 孔板中培养至 70%汇合度。加入 10 μL EdU 溶液并孵育 2 小时后,细胞用 4%多聚甲醛固定。洗涤后,使用 Click-iTR EdU 试剂盒检测 EdU。细胞核用 DAPI 染色,并使用荧光显微镜(奥林巴斯)观察。所有实验均进行三次重复。
2.13. TUNEL assays 2.13. TUNEL 检测
Cells were grown in 24‐well plates until 70% confluence. Cultured cells were fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton‐X 100. TUNEL assays were performed according to the manufacturer's instructions (Roche). Briefly, cells were first incubated in a terminal deoxynucleotidyl transferase (TdT) reaction cocktail, followed by treatment with a Click‐iT reaction cocktail. Nuclei were stained with DAPI. The cells were observed and imaged using a fluorescence microscope (Olympus). All experiments were performed in triplicates.
细胞在 24 孔板中培养至 70%融合。培养的细胞用 4%多聚甲醛固定,并用 0.25% Triton-X 100 透化。根据制造商的说明(罗氏)进行 TUNEL 检测。简而言之,细胞首先在末端脱氧核苷酸转移酶(TdT)反应混合物中孵育,然后用 Click-iT 反应混合物处理。细胞核用 DAPI 染色。使用荧光显微镜(奥林巴斯)观察和成像细胞。所有实验均进行三次重复。
2.14. Flow cytometry analysis
2.14. 流式细胞术分析
Cells (2 × 106) were seeded into each well of a six‐well plate and incubated for 24 hours. To assess apoptosis, cells were harvested after transfection for flow cytometry analysis. Briefly, after double‐staining with an Annexin V‐FITC/PI apoptosis kit (Multi Sciences), apoptosis was determined using a flow cytometer (BD). The apoptotic cells were gated as Annexin V‐FITC+PI+ and Annexin V‐FITC+PI−. The apoptosis rate was defined as the percentage of apoptotic cells in total cells. Flow cytometric analysis was performed to determine the percentage of Treg cells. The cocultured peripheral blood mononuclear cells (PBMCs) were digested with 0.25% trypsin and washed with PBS containing 0.5% (w/v) bovine serum albumin (BSA). Lymphocytes in PBMCs were gated by forward scattering (FSC) and lateral scattering (SSC). CD4+ T cells were gated by CD3 and CD4 staining. Isolated CD4+ T cells were then incubated with FITC‐conjugated anti‐CD25 and anti‐CD127 antibodies.
30
All experiments were performed in triplicates.
细胞 (2 × 106) 被接种到六孔板的每个孔中,并孵育 24 小时。为了评估凋亡,转染后收集细胞进行流式细胞术分析。简而言之,使用 Annexin V‐FITC/PI 凋亡试剂盒 (Multi Sciences) 进行双重染色后,使用流式细胞仪 (BD) 确定凋亡。凋亡细胞被划分为 Annexin V‐FITC+PI+ 和 Annexin V‐FITC+PI−。凋亡率定义为凋亡细胞在总细胞中的百分比。进行流式细胞术分析以确定 Treg 细胞的百分比。共培养的外周血单核细胞 (PBMCs) 用 0.25% 胰蛋白酶消化,并用含有 0.5% (w/v) 牛血清白蛋白 (BSA) 的 PBS 洗涤。PBMCs 中的淋巴细胞通过前向散射 (FSC) 和侧向散射 (SSC) 进行划分。CD4+ T 细胞通过 CD3 和 CD4 染色进行划分。分离的 CD4+ T 细胞随后与 FITC 标记的抗 CD25 和抗 CD127 抗体孵育。 30 所有实验均以三次重复进行。
2.15. Transwell assays 2.15. Transwell 实验
After transfection, 2 × 105 cells were seeded into each well of the upper transwell chamber (8 pm pore size, Corning). A medium containing 10% FBS was added to the lower chamber. After incubation for 24 hours, cells on the upper side of the upper chamber were wiped off, and cells on the lower side of the upper chamber were fixed with 4% paraformaldehyde and stained with 5% crystal violet for 5 minutes. Finally, stained cells were counted under a microscope in five random visual fields. All experiments were performed in triplicates.
转染后,将 2 × 105个细胞接种到上层透析室的每个孔中(孔径 8 pm,Corning)。在下层室中添加含有 10% FBS 的培养基。经过 24 小时的孵育后,擦去上层室上侧的细胞,用 4%多聚甲醛固定下层室上侧的细胞,并用 5%结晶紫染色 5 分钟。最后,在显微镜下在五个随机视野中计数染色细胞。所有实验均进行三次重复。
2.16. Wound‐healing assays
2.16. 创伤愈合实验
Cells were seeded into six‐well plates (2 × 106/well) and incubated for 24 hours. Wounds were created by passing a plastic tip across the monolayer cells. The time of wound infliction was considered to be 0 hours, and wound closure was photographed 24 hours later using a microscope. All experiments were performed in triplicates.
细胞被接种到六孔板中(2 × 106/孔),并孵育 24 小时。通过用塑料尖端划过单层细胞来创建伤口。伤口造成的时间被认为是 0 小时,伤口闭合的照片在 24 小时后使用显微镜拍摄。所有实验均进行三次重复。
2.17. Coculture of PBMCs and PCa cells
2.17. PBMCs 与前列腺癌细胞的共培养
Peripheral blood mononuclear cells obtained from Procell (Wuhan) were cultured for 72 hours for activation in RPMI‐1640 medium (Hyclone; GE Healthcare) with 10% FCS (Gibco; Thermo Fisher Scientific, Inc.), 2 mm l‐glutamine, 0.5% streptomycin and penicillin, 25 mm HEPES, 3 μG/mL anti‐CD28 antibody, 1 μG/mL anti‐CD3 antibody, and 100 u/mL IL‐2. PCa cells (PC3 or DU145) with or without HNRNPC interference were cocultured indirectly with activated PBMCs in six‐well transwell coculture plates (0.4 μm polyester film) for 48 hours, where PBMCs cells were planted in the upper layer and PCa cells were plated in the lower layer.
从 Procell(武汉)获得的外周血单核细胞在 RPMI‐1640 培养基(Hyclone;GE Healthcare)中培养 72 小时以进行激活,培养基中含有 10% FCS(Gibco;Thermo Fisher Scientific, Inc.)、2 mm l‐谷氨酰胺、0.5%链霉素和青霉素、25 mm HEPES、3 μG/mL 抗 CD28 抗体、1 μG/mL 抗 CD3 抗体和 100 u/mL IL‐2。带或不带HNRNPC干扰的 PCa 细胞(PC3 或 DU145)与激活的 PBMCs 在六孔转移培养板(0.4 μm 聚酯膜)中间接共培养 48 小时,其中 PBMCs 细胞种植在上层,PCa 细胞种植在下层。
2.18. Statistical analyses
2.18. 统计分析
All analyses were performed using R software (version 4.0.5). Differences between groups were evaluated using Wilcoxon rank‐sum tests for continuous data and Fisher's exact tests for categorical variables. Pearson's test was used for the correlation analysis. The R package “ConsensusClusterPlus” was used for unsupervised clustering. ssGSEA scores were calculated using the “GSVA” package. K‐M plots were constructed using the K‐M “survival” package. The immune score and the tumor purity were calculated with the “estimate” package based on gene expression levels. CIBERSORT and ImmuCellAI (http://bioinfo.life.hust.edu.cn/ImmuCellAI) were used to calculate the infiltration score, assess the response to immunotherapy, and estimate the abundance of immune cells. All analyses were two‐sided, and statistical significance was set at p < 0.05.
所有分析均使用 R 软件(版本 4.0.5)进行。组间差异使用 Wilcoxon 秩和检验评估连续数据,使用 Fisher 精确检验评估分类变量。相关性分析使用 Pearson 检验。R 包“ConsensusClusterPlus”用于无监督聚类。ssGSEA 分数使用“GSVA”包计算。K-M 图使用 K-M “survival”包构建。免疫评分和肿瘤纯度基于基因表达水平使用“estimate”包计算。CIBERSORT 和 ImmuCellAI(http://bioinfo.life.hust.edu.cn/ImmuCellAI)用于计算浸润评分,评估对免疫治疗的反应,并估计免疫细胞的丰度。所有分析均为双侧,统计显著性设定为 p < 0.05。
3. RESULTS 3. 结果
3.1. Identification of m6A‐dependent IMSs
3.1. m6A 依赖的 IMSs 的识别
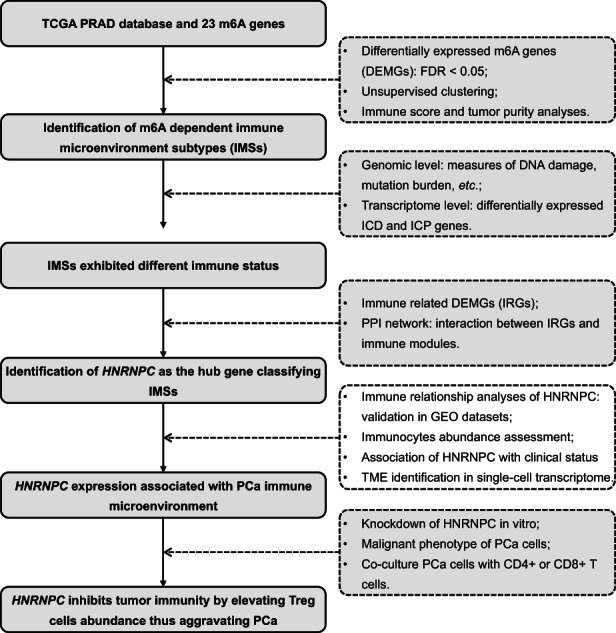
We extracted the m6A regulatory gene (shown in Figure S1) expression profile data of 52 normal and 395 tumor tissues from TCGA. Eleven m6A genes were found to be differentially distributed between PCa and normal tissues with an FDR < 0.05, including RBM15B, IGF2BP2, FMR1, HNRNPA2B1, HNRNPC, METTL3, ZC3H13, YTHDF1, FTO, YTHDF2, and ALKBH5 (Figure 2A). Using unsupervised clustering based on 11 identified enzymes, PCa patients were separated into two clusters with different molecular and immune characteristics (Figure 2B). The baseline characteristics of the 203 group1 and 292 group2 patients are shown in Table S2. After excluding individuals with missing values, the heatmap is shown in Figure 2C, which shows the distributions of tumor stage, BCR condition, Gleason score, m6A gene expression, tumor purity, and immune score between subgroups. Group2 exhibited an abundance enrichment of m6A genes. Furthermore, patients in group2 had higher tumor purity and lower immune scores (Figure 2D,E). Therefore, we named the two subgroups immune microenvironment subtype 1 (IMS1) and IMS2 in the further study. Additionally, we found that patients with IMS2 had a more advanced tumor condition and poorer prognosis than those with IMS1 (Figure 2C,G,H). More importantly, IMS1 samples showed significantly higher scores for both stimulatory and inhibitory immune responses than IMS2 samples (p
all < 0.05, Figure 2F), indicating that IMS1 is an immune “hot” phenotype, while IMS2 is an immune “cold” phenotype. All above mentioned suggested that the enrichment of m6A gene may affect the reprogramming of tumor immune microenvironment, thus leading to different prognoses and survival outcomes.
我们提取了来自 TCGA 的 52 个正常组织和 395 个肿瘤组织的 m6A 调控基因(如图S1)表达谱数据。发现 11 个 m6A 基因在前列腺癌(PCa)和正常组织之间存在差异分布,FDR < 0.05,包括RBM15B、IGF2BP2、FMR1、HNRNPA2B1、HNRNPC、METTL3、ZC3H13、YTHDF1、FTO、YTHDF2和ALKBH5(图2A)。基于 11 个已识别酶的无监督聚类,PCa 患者被分为两个具有不同分子和免疫特征的簇(图2B)。203 名组 1 和 292 名组 2 患者的基线特征见表S2。在排除缺失值的个体后,热图如图2C所示,显示了亚组之间肿瘤分期、BCR 状态、Gleason 评分、m6A 基因表达、肿瘤纯度和免疫评分的分布。组 2 表现出 m6A 基因的丰度富集。 此外,组 2 的患者肿瘤纯度更高,免疫评分更低(图 2D,E)。因此,我们在进一步研究中将这两个亚组命名为免疫微环境亚型 1(IMS1)和 IMS2。此外,我们发现 IMS2 患者的肿瘤状况更为严重,预后也比 IMS1 患者差(图 2C,G,H)。更重要的是,IMS1 样本的刺激性和抑制性免疫反应评分显著高于 IMS2 样本(pall < 0.05,图 2F),这表明 IMS1 是免疫“热”表型,而 IMS2 是免疫“冷”表型。以上所有结果表明,m6A 基因的富集可能影响肿瘤免疫微环境的重编程,从而导致不同的预后和生存结果。
FIGURE 2. 图 2。
DEMGs and cluster analysis. A, The bar plot of 11 DEMGs. B, Unsupervised clustering of PCa patients from TCGA PRAD dataset. C, The heatmap plot of differences in the clinical M stage, pathologic T stage, pathologic N stage, Gleason score, incidence of biochemical recurrence (BCR), tumor purity, and immune score between two clusters. D, E, Differences in the tumor purity and the immune score between two clusters. F, Differences in the state of tumor immune microenvironment, including stimulatory (left) and inhibitory (right) immune scores, between two clusters. G, Kaplan‐Meier plot of the comparison in the overall survival between two clusters. H, The number of patients in different clusters stratified by survival years. *p < 0.05; **p < 0.01; ***p < 0.001. DEMGs, differentially expressed m6A genes; PCa, prostate cancer; PRAD, prostate adenocarcinoma; TCGA, The Cancer Genome Atlas
DEMGs 和聚类分析。A,11 个 DEMGs 的条形图。B,来自 TCGA PRAD 数据集的 PCa 患者的无监督聚类。C,两个簇之间临床 M 期、病理 T 期、病理 N 期、Gleason 评分、生化复发 (BCR) 发生率、肿瘤纯度和免疫评分差异的热图。D,E,两个簇之间肿瘤纯度和免疫评分的差异。F,两个簇之间肿瘤免疫微环境状态的差异,包括刺激性(左)和抑制性(右)免疫评分。G,两个簇之间总体生存率比较的 Kaplan-Meier 图。H,按生存年限分层的不同簇中的患者数量。*p < 0.05; **p < 0.01; ***p < 0.001。DEMGs,差异表达的 m6A 基因;PCa,前列腺癌;PRAD,前列腺腺癌;TCGA,癌症基因组图谱
3.2.
IMSs exhibited different immune status on genomic and transcriptome levels
3.2. IMS 在基因组和转录组水平上表现出不同的免疫状态
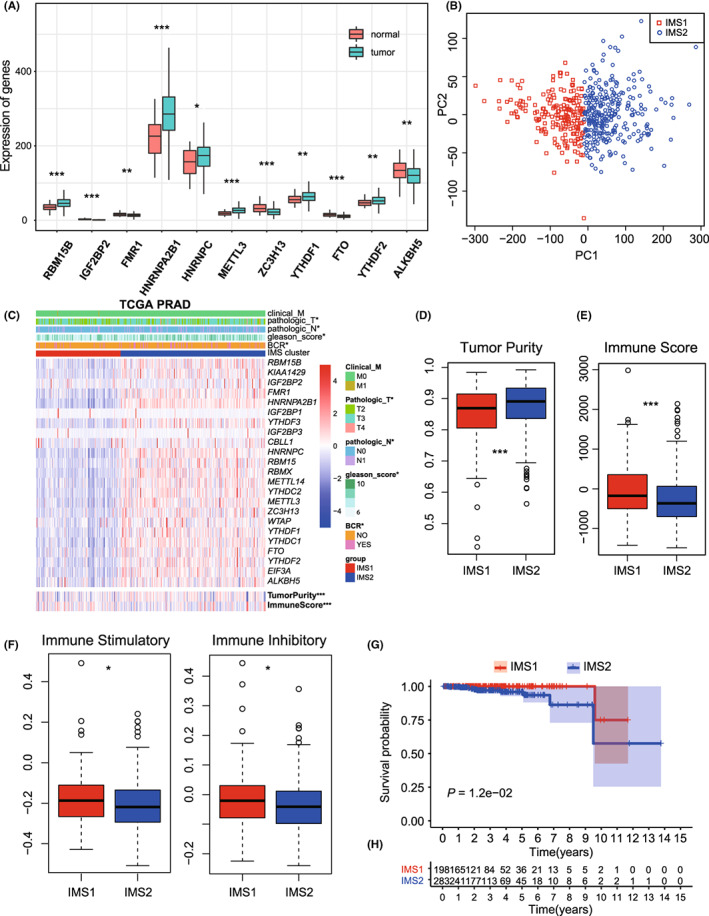
The measures of DNA damage, including altered fraction, AS, HRD, ITH, and number of segments, were all significantly stronger in IMS2 than in IMS1, which represents a poorer prognosis of tumor patients (p
all < 0.05, Figure 3A–E). The mutation rates were also found to be significantly different between IMSs (p
all < 0.05, Figure 3F,G). Further comparison of the mutation spectra revealed several high‐frequency mutations. For example, LRP1B and RYR2 were specifically mutated in IMS1, while SPTA1, ATM, and CSMD3 were mutated in IMS2 (Figure S2A,B). Notable difference was not found in the percentage of immune subtypes (C1‐C4), neoantigen counts (both Indel and SNV), BCR/TCR diversity, and TMB between IMSs (Figures S3 and S4A).
DNA 损伤的测量,包括改变的比例、AS、HRD、ITH 和片段数量,在 IMS2 中均显著强于 IMS1,这代表了肿瘤患者较差的预后(pall < 0.05,图3A–E)。突变率在 IMS 之间也发现显著差异(pall < 0.05,图3F,G)。进一步比较突变谱揭示了几种高频突变。例如,LRP1B 和 RYR2 在 IMS1 中特异性突变,而 SPTA1、ATM 和 CSMD3 在 IMS2 中突变(图S2A,B)。在免疫亚型(C1-C4)、新抗原计数(包括 Indel 和 SNV)、BCR/TCR 多样性和 TMB 之间未发现显著差异(图S3和S4A)。
FIGURE 3. 图 3。
Immune landscape of immune microenvironment subtype (IMS) clusters. A‐E, Measures of DNA damage, fraction altered, number of segments, aneuploidy score (AS), homologous recombination defects (HRD), intratumor heterogeneity (ITH), between clusters. F, G, silent and nonsilent mutation rate of IMS clusters. H, Correlation between the expression level of HNRNPC and immune score in TCGA database. I, Protein‐protein interaction (PPI) network of the interactions between immune‐related genes (IRGs) and major histocompatibility complex (MHC) module. J, Correlations of HNRNPC expression with MEX3B expression in TCGA database
免疫微环境亚型(IMS)簇的免疫景观。A‐E,DNA 损伤的测量、改变的比例、片段数量、非整倍体评分(AS)、同源重组缺陷(HRD)、肿瘤内异质性(ITH)在簇之间的比较。F,G,IMS 簇的沉默和非沉默突变率。H,HNRNPC的表达水平与 TCGA 数据库中的免疫评分之间的相关性。I,免疫相关基因(IRGs)与主要组织相容性复合体(MHC)模块之间相互作用的蛋白质-蛋白质相互作用(PPI)网络。J,HNRNPC表达与 TCGA 数据库中 MEX3B 表达的相关性。
The expression of most ICD and ICP genes varied significantly between IMSs (Figure S3D,E), indicating a difference in the tumor immune status. Most importantly, well‐established immunotherapy markers showed obvious differences. CD274 (PD‐L1),
21
T‐cell immunoglobulin mucin‐3 (TIM‐3),
22
and CTLA4,
23
which represent the most effective predictive biomarkers for checkpoint inhibitor–based immunotherapy, were expressed at higher levels in IMS2 (p
all < 0.05, Figure S2C). CXCL9, the ligand of CXCR3, was also expressed at higher levels in IMS2 cells (Figure S2C, p = 1.94 × 10−9). CXCL9 has been identified as a biomarker for sensitivity to PD‐1 blockade, and augmenting the intratumoral function of this CXCR3‐CXCL9 chemokine system could improve clinical outcomes.
24
MEX3B was also significantly higher in IMS2 cells (p = 2.7 × 10−14), which can inhibit the killing effect by preventing T cells from recognizing tumor cells.
25
Furthermore, tumors treated with PD‐1/PD‐L1–blocking antibodies developed drug resistance through the upregulation of CD38,
26
which showed higher expression in IMS1 (p = 5.43 × 10–3, Figure S2C).
大多数 ICD 和 ICP 基因的表达在 IMS 之间显著不同(图S3D,E),这表明肿瘤免疫状态存在差异。最重要的是,已确立的免疫治疗标志物显示出明显差异。CD274(PD-L1), 21 T 细胞免疫球蛋白粘蛋白-3(TIM-3), 22 和 CTLA4, 23 作为基于检查点抑制剂的免疫治疗最有效的预测生物标志物,在 IMS2 中的表达水平更高(pall < 0.05,图S2C)。CXCL9,CXCR3 的配体,在 IMS2 细胞中的表达水平也更高(图S2C,p = 1.94 × 10−9)。CXCL9 已被确定为对 PD-1 阻断敏感性的生物标志物,增强这一 CXCR3-CXCL9 趋化因子系统的肿瘤内功能可能改善临床结果。 24 MEX3B 在 IMS2 细胞中的表达也显著更高(p = 2.7 × 10−14),它可以通过防止 T 细胞识别肿瘤细胞来抑制杀伤效应。 25 此外,接受 PD-1/PD-L1 阻断抗体治疗的肿瘤通过上调 CD38 发展出药物耐药性, 26 其在 IMS1 中的表达更高 (p = 5.43 × 10–3, 图 S2C)。
3.3. Identification of HNRNPC as the hub gene classifying IMSs
3.3. 确定 HNRNPC 作为分类 IMS 的中心基因
To investigate the crosstalk between m6A modification and the immune microenvironment, we analyzed the immune correlation of 11 DEMGs. Correlation analysis identified six IRGs, HNRNPC, HNRNPA2B1, YTHDF1, YTHDF2, METTL3, and RBM15B, whose expression levels were significantly negatively correlated with the immune score (R = −0.19, −0.18, −0.15, −0.13, −0.28, and −0.23, respectively; p = 2.4 × 10−5, 5.3 × 10−5, 7.2 × 10−4, 4.0 × 10−3, 1.8 × 10−10, and 1.6 × 10−7, respectively; Figure 3H and Figure S4B–F) and positively correlated with tumor purity (R = 0.20, 0.19, 0.17, 0.16, 0.29, and 0.23, respectively; p = 4.8 × 10−6, 2.5 × 10−5, 1.3 × 10−4, 2.6 × 10−4, 4.0 × 10−11, and 1.3 × 10−7, respectively; Figure S4G–L), suggesting that these genes may be associated with a low degree of immune invasion and tumor progression (Figure S5). In addition, PPI network analysis was performed to identify hub regulatory factors. As HNRNPC was associated with the most nodes, it was identified as a hub gene, which also showed association with the MHC module simultaneously (Figure 3I). The PPI network with the immunoinhibitory and immunostimulatory modules also confirmed the immune correlation of m6A regulatory factors (Figure S6). HNRNPC was also significantly upregulated in IMS2 (Figure 2C); therefore, we noted it as the representative classifying IMS in further studies.
为了研究 m6A 修饰与免疫微环境之间的相互作用,我们分析了 11 个差异表达基因(DEMGs)的免疫相关性。相关性分析确定了六个免疫相关基因(IRGs),HNRNPC、HNRNPA2B1、YTHDF1、YTHDF2、METTL3 和 RBM15B,其表达水平与免疫评分显著负相关(R = −0.19、−0.18、−0.15、−0.13、−0.28 和−0.23,分别;p = 2.4 × 10−5、5.3 × 10−5、7.2 × 10−4、4.0 × 10−3、1.8 × 10−10和 1.6 × 10−7,分别;图3H和图S4B–F)并与肿瘤纯度显著正相关(R = 0.20、0.19、0.17、0.16、0.29 和 0.23,分别;p = 4.8 × 10−6、2.5 × 10−5、1.3 × 10−4、2.6 × 10−4、4.0 × 10−11和 1.3 × 10−7,分别;图S4G–L),这表明这些基因可能与低程度的免疫侵袭和肿瘤进展相关(图S5)。 此外,进行了 PPI 网络分析以识别中心调控因子。由于HNRNPC与最多节点相关,因此被确定为中心基因,同时也显示出与 MHC 模块的关联(图3I)。具有免疫抑制和免疫刺激模块的 PPI 网络也确认了 m6A 调控因子的免疫相关性(图S6)。HNRNPC在 IMS2 中也显著上调(图2C);因此,我们在进一步研究中将其视为代表性分类 IMS。
3.4.
HNRNPC expression associated with PCa immune microenvironment
3.4. HNRNPC 表达与前列腺癌免疫微环境相关
To investigate the effect of the hub m6A regulator HNRNPC on the tumor immune microenvironment of PCa, four GEO datasets were used for validation. Consistent results were obtained for the different datasets. In the GSE46602, GSE32571, and GSE60329 datasets, HNRNPC showed a negative correlation with immune score, but a positive correlation with tumor purity (Figure S7). In dataset GSE70768 with the largest sample size, the same result was obtained with statistical significance (R = −0.20 and 0.24, respectively; p = 5.3 × 10−3 and 5.9 × 10−4, respectively; Figure S7A). Additionally, the expression level of HNRNPC was negatively correlated with the infiltration score (R = −0.34; p = 8.7 × 10−7; Figure S7C). Meanwhile, we found that the expression of HNPNPC was significantly correlated with MEX3B (R = 0.40, p < 2.2 × 10−16; Figure 3J). Finally, the correlation between HNRNPC and the immune cells was investigated. A high abundance of natural regulatory T cells (nTreg), induced regulatory T cells (iTreg), T helper 1 cells (Th1), central memory cells, and monocytes could be identified in the HNRNPC low‐expression group (p
all < 0.05, Figure 4A). Overexpression of Treg cells is likely to be the reason for the tumor's immunosuppressive status. Meanwhile, the abundance of exhausted cells, natural killer T cells (NKT), and natural killer cells (NK) was significantly lower in the high‐expression group (p
all < 0.05, Figure 4A). To verify the relationship between HNRNPC expression and abundant of Treg, cytotoxic T cells, we performed a correlation analysis in the TCGA‐PRAD cohort, finding that the expression of HNRNPC was significantly positively correlated with Treg (iTreg and nTreg) scores, and negatively with cytotoxic T cell scores (R = 0.1, 0.31 and 0.11, respectively, p = 2.2 × 10−2, 1.1 × 10−12 and 1.1 × 10−2, respectively; Figure 4B–D). Furthermore, HNRNPC was highly expressed in progressive PCa, such as higher pathologic T stage, higher pathologic N stage, and high Gleason score (Figure 4E–H) in the TCGA PRAD dataset. In addition, the OS of patients with high HNRNPC expression was poorer than that of patients with low expression (Figure 4I).
为了研究中心 m6A 调节因子HNRNPC对前列腺癌(PCa)肿瘤免疫微环境的影响,使用了四个 GEO 数据集进行验证。不同数据集得到了相似的结果。在GSE46602、GSE32571和GSE60329数据集中,HNRNPC与免疫评分呈负相关,但与肿瘤纯度呈正相关(图S7)。在样本量最大的GSE70768数据集中,得到了相同的结果,并具有统计学意义(R = −0.20 和 0.24;p = 5.3 × 10−3和 5.9 × 10−4;图S7A)。此外,HNRNPC的表达水平与浸润评分呈负相关(R = −0.34;p = 8.7 × 10−7;图S7C)。同时,我们发现 HNPNPC 的表达与 MEX3B 显著相关(R = 0.40,p < 2.2 × 10−16;图3J)。 最后,研究了HNRNPC与免疫细胞之间的相关性。在HNRNPC低表达组中,可以识别出高丰度的自然调节性 T 细胞(nTreg)、诱导性调节性 T 细胞(iTreg)、T 辅助细胞 1(Th1)、中心记忆细胞和单核细胞(pall < 0.05,图4A)。Treg 细胞的过表达可能是肿瘤免疫抑制状态的原因。同时,高表达组中耗竭细胞、自然杀伤 T 细胞(NKT)和自然杀伤细胞(NK)的丰度显著降低(pall < 0.05,图4A)。为了验证HNRNPC表达与 Treg、细胞毒性 T 细胞丰度之间的关系,我们在 TCGA-PRAD 队列中进行了相关性分析,发现HNRNPC的表达与 Treg(iTreg 和 nTreg)评分显著正相关,而与细胞毒性 T 细胞评分显著负相关(R = 0.1,0.31 和 0.11,分别为p = 2.2 × 10−2,1.1 × 10−12和 1。1 × 10−2,分别;图4B–D)。此外,HNRNPC 在进展性前列腺癌中高度表达,例如更高的病理 T 分期、更高的病理 N 分期和高 Gleason 评分(图4E–H)在 TCGA PRAD 数据集中。此外,高HNRNPC表达的患者的生存期比低表达患者差(图4I)。
FIGURE 4. 图 4。
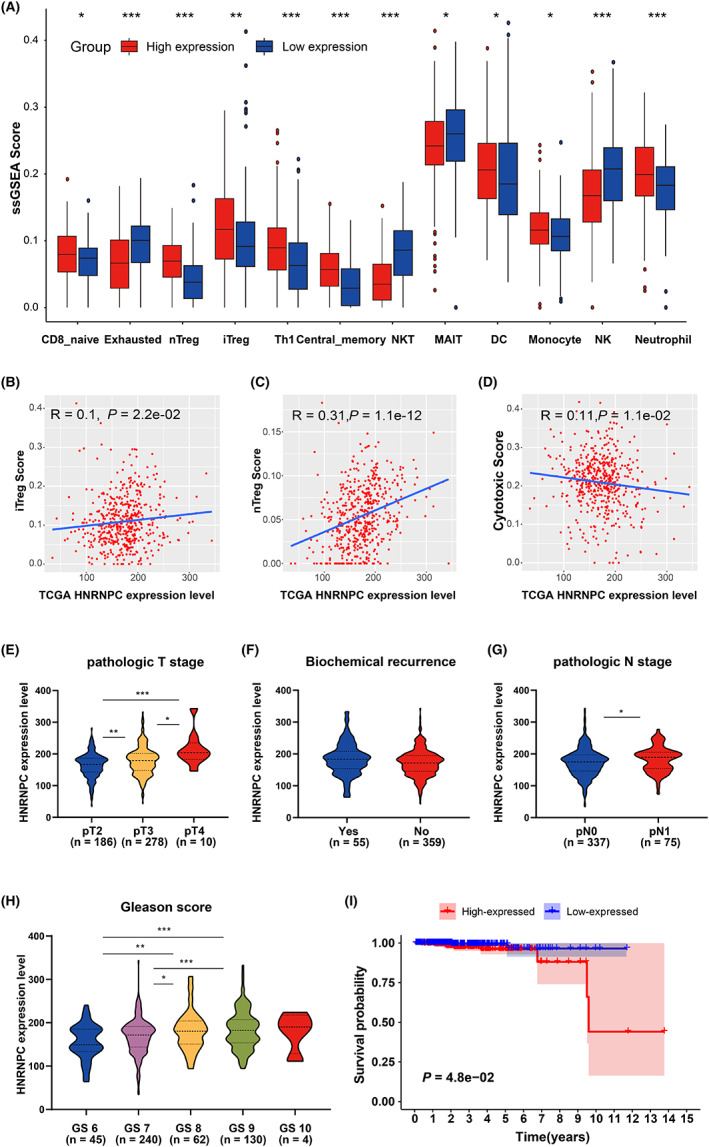
Validation of the correlation between HNRNPC and immune microenvironment. A, Differences in the abundance of immune cells between the high‐expression group and low‐expression group (separated according to the median of HNRNPC expression level in GSE70768). *p < 0.05. B‐D, Correlations between HNRNPC expression and iTreg (B), nTreg (C), and cytotoxic T cells score (D). E‐H, Expression difference of HNRNPC between tumor patients with different pathologic T stages (E), biochemical recurrence condition (F), pathologic N stage (G), and Gleason score (H). I, Kaplan‐Meier plot of the comparison in the overall survival between HNRNPC high‐expression patients and low‐expression patients. TCGA, The Cancer Genome Atlas; PRAD, prostate adenocarcinoma
验证HNRNPC与免疫微环境之间的相关性。A、高表达组与低表达组之间免疫细胞丰度的差异(根据GSE70768中HNRNPC表达水平的中位数分组)。*p < 0.05。B-D、HNRNPC表达与 iTreg(B)、nTreg(C)和细胞毒性 T 细胞评分(D)之间的相关性。E-H、不同病理 T 分期(E)、生化复发情况(F)、病理 N 分期(G)和 Gleason 评分(H)下肿瘤患者的HNRNPC表达差异。I、HNRNPC高表达患者与低表达患者在总体生存率比较的 Kaplan-Meier 图。TCGA,癌症基因组图谱;PRAD,前列腺腺癌
3.5. Treg proportion in HNRNPC‐positive cells was significantly higher than in negative cells on the single‐cell level
3.5. HNRNPC 阳性细胞中的 Treg 比例在单细胞水平上显著高于阴性细胞。
To accurately map T cell status on the single‐cell level, we described the single‐cell transcriptome atlas of PCa. First, after data preprocessing, we obtained a total of 36,424 cells (Figure 5A,B and Figure S8A). We annotated nine cell types: luminal cells (AR), T cells (KRT3E), B cells (MS4A1), mast cells (KIT), monocytic cells (CD14), endothelial (CD34), fibroblast (ACAT2), basal (KRT19), and other epithelial cells (Figure S8B). Furthermore, we analyzed the correlation between the proportion of each cell type and HNRNPC‐positive cell rate, the result showed that HNRNPC‐positive cell rate was significantly positively correlated with epithelium cells, and negatively with T cells (Figure 5E). Through subanalyses on T cells, we got nine clusters including CD4_NaiveLike, CD8_EarlyActive, CD8_EffectorMemory, CD8_NaiveLike, CD8_Tex, CD8_Tpex, Tfh, Th1, and Treg. According to the expression of HNRNPC, we separated the cells into HNRNPC‐positive and ‐negative cells. The proportion of Treg cells in positive cells was much higher than that in HNRNPC‐negative cells. Furthermore, the number of naïve CD8 T cells and active CD8 T cells was lower in HNRNPC‐positive cells, which indicates that cells with high expression of HNRNPC were in a state of immunosuppression (Figure 5D–F).
为了准确映射单细胞水平的 T 细胞状态,我们描述了前列腺癌的单细胞转录组图谱。首先,在数据预处理后,我们获得了总共 36,424 个细胞(图 5A,B 和图 S8A)。我们注释了九种细胞类型:腔道细胞(AR)、T 细胞(KRT3E)、B 细胞(MS4A1)、肥大细胞(KIT)、单核细胞(CD14)、内皮细胞(CD34)、成纤维细胞(ACAT2)、基底细胞(KRT19)和其他上皮细胞(图 S8B)。此外,我们分析了每种细胞类型的比例与 HNRNPC 阳性细胞率之间的相关性,结果显示 HNRNPC 阳性细胞率与上皮细胞显著正相关,而与 T 细胞显著负相关(图 5E)。通过对 T 细胞的亚分析,我们得到了九个簇,包括 CD4_NaiveLike、CD8_EarlyActive、CD8_EffectorMemory、CD8_NaiveLike、CD8_Tex、CD8_Tpex、Tfh、Th1 和 Treg。根据 HNRNPC 的表达,我们将细胞分为 HNRNPC 阳性和阴性细胞。阳性细胞中 Treg 细胞的比例远高于 HNRNPC 阴性细胞中的比例。 此外,HNRNPC阳性细胞中天真 CD8 T 细胞和活跃 CD8 T 细胞的数量较低,这表明高表达HNRNPC的细胞处于免疫抑制状态(图5D–F)。
FIGURE 5. 图 5。
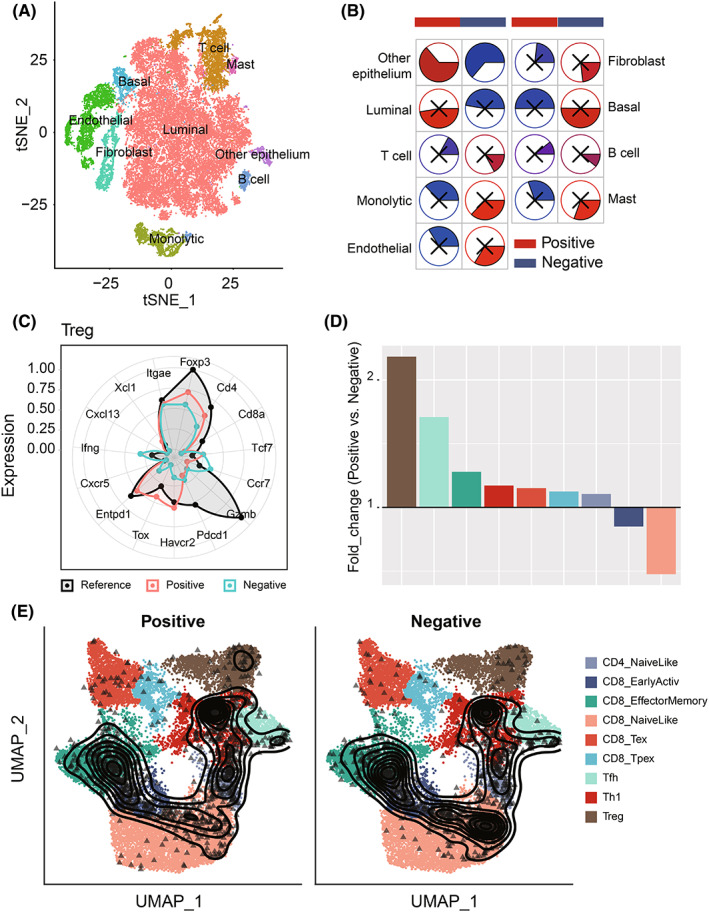
Comparison of Treg proportion in HNRNPC‐positive cells and ‐negative cells on the single‐cell level. A, The tSNE clustering visualization of single‐cell RNA sequencing of prostate cancer. B, The tSNE plots of cells from patients with cells colored based on the cell types. C, Well‐known markers of eight cell types. D, The fold change of T cell subset composition of positive and negative cells. E, The tSNE plot of T cells colored by subtype and HNRNPC expression.
在单细胞水平上比较HNRNPC‐阳性细胞和阴性细胞中的 Treg 比例。A,前列腺癌单细胞 RNA 测序的 tSNE 聚类可视化。B,基于细胞类型对患者细胞着色的 tSNE 图。C,八种细胞类型的知名标记物。D,阳性和阴性细胞的 T 细胞亚群组成的倍数变化。E,按亚型和HNRNPC表达着色的 T 细胞 tSNE 图。
Furthermore, we explored specific cell‐cell communication in positive samples. According to the cutoff value of the positive rate of 70%, we divided 13 samples into two groups: positive group (five samples) and negative group (eight samples) (Figure S9A). The differentially enriched interaction between cell types is shown in the heatmap, which indicated that the cell‐cell interaction in the positive group was stronger than that in the negative group, especially between T cells and other cells (Figure S9B). Then, different signal flow analyses implied that a large number of signal flows were activated in the positive group, including SPP1, TGF‐β, which were associated with immunosuppression (Figure S9C). Finally, we discovered ligand‐receptor interaction specifically in the HNRNPC‐positive group. The epithelial cells in the HNRNPC‐positive group interacted with other types of cells through such ligand‐receptor pairs: MIF‐(CD74+CXCR4) and MIF‐(CD74+CD44) with T cells; VEGFR‐PDGFA (a tumor angiogenesis related interaction), and NAMPT‐(ITGA5+ITGB1) (an immunosuppression‐associated interaction) with endothelial cells, and JAG1‐NOTCH3 and PDGFA‐PDGFRB (two pairs associated with pericytes and formation of tumor‐associated fibroblasts) with fibroblast (Figure S9D).
此外,我们探讨了阳性样本中特定的细胞间通信。根据 70%的阳性率截止值,我们将 13 个样本分为两组:阳性组(五个样本)和阴性组(八个样本)(图 S9A)。细胞类型之间的差异富集相互作用在热图中显示,阳性组的细胞间相互作用强于阴性组,尤其是在 T 细胞与其他细胞之间(图 S9B)。然后,不同的信号流分析暗示阳性组中激活了大量信号流,包括与免疫抑制相关的 SPP1 和 TGF-β(图 S9C)。最后,我们在 HNRNPC-阳性组中特别发现了配体-受体相互作用。 在HNRNPC阳性组中,上皮细胞通过以下配体-受体对与其他类型的细胞相互作用:MIF-(CD74+CXCR4)和 MIF-(CD74+CD44)与 T 细胞;VEGFR-PDGFA(与肿瘤血管生成相关的相互作用),以及 NAMPT-(ITGA5+ITGB1)(与免疫抑制相关的相互作用)与内皮细胞,以及 JAG1-NOTCH3 和 PDGFA-PDGFRB(与周细胞和肿瘤相关成纤维细胞形成相关的两个配对)与成纤维细胞(图S9D)。
3.6. Knockdown of HNRNPC inhibited the proliferation and migration ability of PCa cell in vitro
3.6. HNRNPC 的敲除抑制了前列腺癌细胞在体外的增殖和迁移能力
We performed PCR analysis to confirm the expression levels of HNRNPC in PCa cell lines. Compared with the normal prostate epithelial cell line RWPE‐1, four PCa cell lines (C4‐2B, Lncap, DU145, and PC3) showed elevated HNRNPC expression (Figure 6A). To evaluate the oncogenic effect of HNRNPC in vitro, DU145 and PC3 cells with the highest HNRNPC levels were stably transfected with an shRNA targeting HNRNPC, whereas cells transfected with a scrambled vector were used as a negative control. Using shRNAs, the expression of HNRNPC was successfully knocked down in two PCa cell lines (DU145 and PC3) (Figure 6B, Figure S10A). Among the shRNAs, shRNA‐1 and shRNA‐2 were used for functional assays with the highest knockdown efficiency. We investigated the effect of HNRNPC on PCa cell proliferation. CCK‐8 assay demonstrated that knockdown of HNRNPC significantly decreased the proliferative ability of DU145 and PC3 cells (Figure 6C, Figure S10B). These results were further confirmed by EdU incorporation assays in both DU145 and PC3 cells (Figure 6D, Figure S10C). These results indicated that HNRNPC is required for PCa cell proliferation in vitro. Furthermore, we evaluated the effect of HNRNPC knockdown on apoptosis of PCa cells using TUNEL staining and flow cytometry. Our results showed that the apoptosis level of PCa cells was significantly decreased by HNRNPC downregulation (Figure 6 E‐F, Figure S10D,E). Transwell and wound‐healing assays were performed to detect the effects of HNRNPC on the migration and invasion of PCa cells. The results showed that HNRNPC could also promote malignancy in PCa cells, as evidenced by the decreased migration and invasion ability in DU145 and PC3 cells after knockdown of HNRNPC (Figure 6G, Figure S10F,G).
我们进行了 PCR 分析,以确认在 PCa 细胞系中HNRNPC的表达水平。与正常前列腺上皮细胞系 RWPE-1 相比,四个 PCa 细胞系(C4-2B、Lncap、DU145 和 PC3)显示出HNRNPC表达升高(图6A)。为了评估HNRNPC在体外的致癌效应,DU145 和 PC3 细胞(具有最高的HNRNPC水平)被稳定转染了针对HNRNPC的 shRNA,而转染了随机载体的细胞则作为阴性对照。使用 shRNA,成功在两个 PCa 细胞系(DU145 和 PC3)中敲低了HNRNPC的表达(图6B,图S10A)。在 shRNA 中,shRNA-1 和 shRNA-2 被用于功能实验,具有最高的敲低效率。我们研究了HNRNPC对 PCa 细胞增殖的影响。CCK-8 实验表明,敲低HNRNPC显著降低了 DU145 和 PC3 细胞的增殖能力(图6C,图S10B)。 这些结果通过在 DU145 和 PC3 细胞中进行 EdU 掺入实验进一步确认(图6D,图S10C)。这些结果表明HNRNPC在体外是前列腺癌细胞增殖所必需的。此外,我们使用 TUNEL 染色和流式细胞术评估了HNRNPC敲低对前列腺癌细胞凋亡的影响。我们的结果显示,前列腺癌细胞的凋亡水平因HNRNPC下调而显著降低(图6 E‐F,图S10D,E)。我们进行了 Transwell 和伤口愈合实验,以检测HNRNPC对前列腺癌细胞迁移和侵袭的影响。结果显示,HNRNPC也可以促进前列腺癌细胞的恶性程度,敲低HNRNPC后,DU145 和 PC3 细胞的迁移和侵袭能力均降低(图6G,图S10F,G)。
FIGURE 6. 图 6。
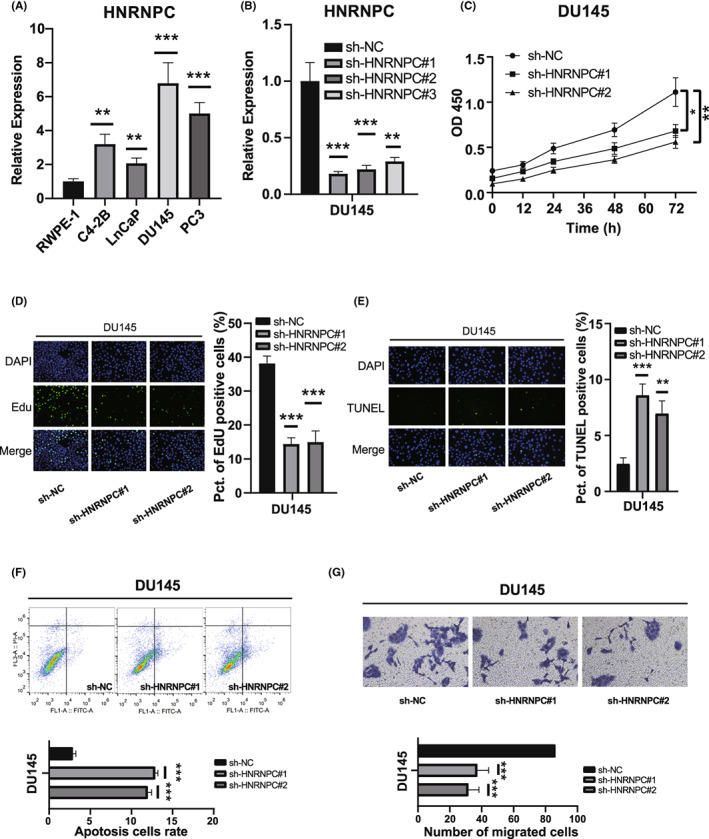
Knockdown of HNRNPC‐promoted cell apoptosis inhibited the cell proliferation and migration of prostate cancer (PCa) cells. A, Relative expression of HNRNPC in human prostate epithelial cell line RWPE‐1 and PCa cell lines. B, qRT‐PCR analyses of HNRNPC mRNA in DU145 cells treated with negative control (NC) or HNRNPC shRNAs. C, D, CCK‐8 assays and EdU incorporation assays for DU145 cells treated with negative control (NC) or HNRNPC shRNAs. E, F, TUNEL assays and flow‐cytometric analysis for DU145 cells treated with NC or HNRNPC shRNAs. G, Transwell assays and wound‐healing assays for DU145 cells
抑制HNRNPC‐的敲除促进了前列腺癌(PCa)细胞的凋亡,抑制了细胞增殖和迁移。A,人体前列腺上皮细胞系 RWPE‐1 和 PCa 细胞系中HNRNPC的相对表达。B,使用阴性对照(NC)或HNRNPC shRNA 处理的 DU145 细胞中HNRNPC mRNA 的 qRT‐PCR 分析。C,D,使用阴性对照(NC)或HNRNPC shRNA 处理的 DU145 细胞的 CCK‐8 实验和 EdU 掺入实验。E,F,使用 NC 或HNRNPC shRNA 处理的 DU145 细胞的 TUNEL 实验和流式细胞术分析。G,DU145 细胞的 Transwell 实验和伤口愈合实验。
3.7.
HNRNPC inhibits tumor immunity by elevating the activation of Treg cells and suppression of CD8 cells
3.7. HNRNPC 通过提高 Treg 细胞的活化和抑制 CD8 细胞来抑制肿瘤免疫
The above immune correlation analysis suggested that HNRNPC could mediate the activation of Treg cells. To determine whether HNRNPC‐mediated PCa cells induced activation of Tregs, we cocultured transfected PCa cells with PBMC T cells (Figure 7A). The expression of the Treg markers (CD25+CD127−) was significantly decreased in PBMC cells incubated with HNRNPC‐knockdown PCa cells compared with that in the NC group (Figure 7B,C), indicating the activation of Treg cells in HNRNPC low‐expression tumors. Furthermore, the number of effector CD8 T cells (CD8+CD107a+IFN‐γ+) was significantly increased in PBMC cells incubated with HNRNPC‐knockdown PCa cells compared with that in the NC group (Figure 7D,E), representing the suppression of CD8 cells.
上述免疫相关分析表明,HNRNPC可能介导 Treg 细胞的激活。为了确定HNRNPC‐介导的 PCa 细胞是否诱导 Tregs 的激活,我们将转染的 PCa 细胞与 PBMC T 细胞共同培养(图7A)。与 NC 组相比,PBMC 细胞中与HNRNPC敲低的 PCa 细胞共同培养后,Treg 标记物(CD25+CD127−)的表达显著降低(图7B,C),这表明在HNRNPC低表达肿瘤中 Treg 细胞的激活。此外,与 NC 组相比,PBMC 细胞中与HNRNPC敲低的 PCa 细胞共同培养后,效应 CD8 T 细胞(CD8+CD107a+IFN‐γ+)的数量显著增加(图7D,E),代表 CD8 细胞的抑制。
FIGURE 7. 图 7。
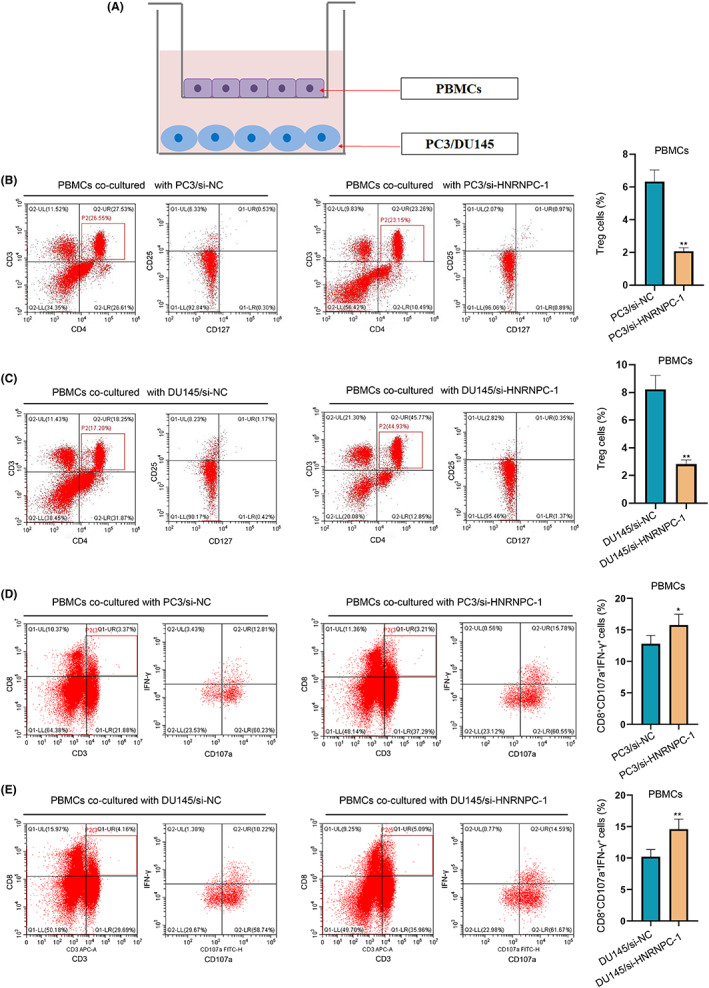
Coculture of peripheral blood mononuclear cells (PBMCs) and prostate cancer (PCa) cells. A, The sketch of the coculture system. B, C, The expression of Treg markers (CD25+CD127−) after coculture‐transfected PC3 cells and DU145 cell. D, E, The expression of effector CD8 T cell markers (CD8+CD107a+INF‐γ+) after coculture‐transfected PC3 cells and DU145 cell
外周血单核细胞(PBMCs)与前列腺癌(PCa)细胞的共培养。A,共培养系统的示意图。B,C,经过共培养转染的 PC3 细胞和 DU145 细胞后,Treg 标记物(CD25+CD127−)的表达。D,E,经过共培养转染的 PC3 细胞和 DU145 细胞后,效应 CD8 T 细胞标记物(CD8+CD107a+INF‐γ+)的表达。
4. DISCUSSION 4. 讨论
Prostate cancer is a heterogeneous disease with different ethnic characteristics that originates from prostatic epithelial cells.
31
,
32
Targeting the immunosuppressive mechanism in the TME has revolutionized cancer treatment, while clinical trials have shown minimal efficacy in patients with PCa. M6A methylation is the most common form of mRNA modification and plays a pivotal role in post‐transcriptional regulation. Studies showed that METTL3 and YTHDF2 promote PCa migration and invasion by mediating m6A modification,
33
,
34
suggesting that m6A regulates the occurrence and development of PCa in a unique way. Furthermore, studies evaluated the m6A regulation patterns of PCa and correlated these modification patterns with the tumor immune microenvironment characteristics.
35
,
36
Although these studies coanalyzed m6A methylation and the TME of PCa, they did not discuss the underlying mechanism of tumor immune status.
前列腺癌是一种异质性疾病,具有不同的种族特征,起源于前列腺上皮细胞。 31 , 32 针对肿瘤微环境中的免疫抑制机制的研究革新了癌症治疗,而临床试验显示前列腺癌患者的疗效有限。M6A 甲基化是最常见的 mRNA 修饰形式,在转录后调控中发挥着关键作用。研究表明,METTL3 和 YTHDF2 通过介导 m6A 修饰促进前列腺癌的迁移和侵袭, 33 , 34 这表明 m6A 以独特的方式调节前列腺癌的发生和发展。此外,研究评估了前列腺癌的 m6A 调控模式,并将这些修饰模式与肿瘤免疫微环境特征相关联。 35 , 36 尽管这些研究共同分析了前列腺癌的 m6A 甲基化和肿瘤微环境,但并未讨论肿瘤免疫状态的潜在机制。
In our study, the expression patterns, prognostic values, and effects on the TME of m6A regulators in PCa were demonstrated. Subsequently, we identified two subtypes of PCa (IMS1 and IMS2) by consensus clustering of m6A regulatory factors. IMS1/IMS2 subtypes have different prognoses and clinicopathological features in PCa patients, which are closely related to immune scores and immune cell infiltration. The prognosis of IMS1 patients was significantly better than that of patients with IMS2, which may be related to higher immune infiltration. This was consistent with previous findings that patients with high immune scores had superior survival compared with those with low immune scores.
37
As most m6A regulators were highly expressed in IMS2 cells, it was suggested that the abundance of m6A modification may remodel the tumor immune microenvironment, leading to an immune “cold” phenotype. At the same time, IMS2 had a higher proportion of DNA damage, such as HRD and ITH. Wang et al showed that a higher HRD score was associated with poor clinical outcomes in PRAD.
38
Several studies have found that ITH can be a prognostic factor for solid tumors, as it diminishes the immune response.
39
,
40
Consistently, the mutation spectrum showed that TP53 and ATM comutations especially occurred in IMS2, which has implications as a biomarker for guiding ICI treatment. TP53 and ATM comutation was associated with better OS than a single mutation and no mutation among patients with any cancer.
41
Most importantly, IMS2 showed higher expression of PD‐L1 and CXCL9, which are all biomarkers of sensitivity to PD‐1 blockade therapy.
42
At the same time, IMS2 cells showed lower expression of CD38, which represented low resistance to anti‐PD‐1 immunotherapy.
26
These results suggest that the IMS2 group probably responded to PD‐1 blockade therapy. However, other ICPs including TIM3 and CTLA4 were also significantly highly expressed in IMS2, which means that the inhibitory immune microenvironment of this group of patients may not be activated by pure antiPD‐1/PD‐L1 therapy. HNRNPC, a m6A reader, was identified as the hub gene interacting with HLA‐A, HLA‐B, and B2M. We found MEX3B, which was proved to mediate resistance to cancer immunotherapy by downregulating HLA‐A expression, showed higher expression in IMS2. Moreover, HNRNPC could regulate the m6A modification of HLA‐A, HLA‐B, and B2M (shown in the RM2Target and RMBase databases
43
,
44
), and a previous study showed that B2M mutations are closely related to the abundance of Treg cells, which might lead to an immunosuppression.
45
In conclusion, a novel immunotherapeutic strategy based on the identification of IMS2 patients might greatly benefit their survival.
在我们的研究中,展示了 m6A 调节因子在前列腺癌(PCa)中的表达模式、预后价值和对肿瘤微环境(TME)的影响。随后,我们通过对 m6A 调节因子的共识聚类识别了两种 PCa 亚型(IMS1 和 IMS2)。IMS1/IMS2 亚型在 PCa 患者中具有不同的预后和临床病理特征,这与免疫评分和免疫细胞浸润密切相关。IMS1 患者的预后显著优于 IMS2 患者,这可能与更高的免疫浸润有关。这与之前的发现一致,即高免疫评分的患者与低免疫评分的患者相比,生存率更高。 37 由于大多数 m6A 调节因子在 IMS2 细胞中高表达,建议 m6A 修饰的丰度可能重塑肿瘤免疫微环境,导致免疫“冷”表型。同时,IMS2 具有更高比例的 DNA 损伤,如 HRD 和 ITH。Wang 等人显示,较高的 HRD 评分与 PRAD 的不良临床结果相关。 38 多项研究发现,ITH 可能是实体肿瘤的预后因素,因为它会削弱免疫反应。 39 , 40 一致地,突变谱显示 TP53 和 ATM 共突变特别发生在 IMS2 中,这为指导 ICI 治疗提供了生物标志物的意义。TP53 和 ATM 共突变与任何癌症患者中单一突变和无突变相比,生存期更好。 41 最重要的是,IMS2 显示出更高的 PD‐L1 和 CXCL9 表达,这些都是对 PD‐1 阻断治疗敏感的生物标志物。 42 与此同时,IMS2 细胞显示出较低的 CD38 表达,代表对抗 PD‐1 免疫治疗的低抵抗性。 26 这些结果表明,IMS2 组可能对 PD‐1 阻断治疗有反应。然而,其他 ICP 包括 TIM3 和 CTLA4 在 IMS2 中也显著高表达,这意味着这一组患者的抑制性免疫微环境可能无法通过单纯的抗 PD‐1/PD‐L1 治疗激活。 HNRNPC,一个 m6A 读取器,被确定为与 HLA‐A、HLA‐B 和 B2M 相互作用的中心基因。我们发现 MEX3B 被证明通过下调 HLA‐A 表达来介导对癌症免疫治疗的抵抗,在 IMS2 中表现出更高的表达。此外,HNRNPC 可以调节 HLA‐A、HLA‐B 和 B2M 的 m6A 修饰(如 RM2Target 和 RMBase 数据库所示 43 , 44 ),而之前的研究表明 B2M 突变与 Treg 细胞的丰度密切相关,这可能导致免疫抑制。 45 总之,基于 IMS2 患者识别的新型免疫治疗策略可能会极大地改善他们的生存。
We identified HNRNPC as a hub gene that interacts with m6A methylation via immune processes and can effectively classify IMSs. Some studies found an association between HNRNPC and tumor progression,
46
,
47
,
48
as well as immune cell infiltration.
49
However, which immune cells can be regulated by HNRNPC, thus remodeling the TME is still unclear.
我们将HNRNPC识别为一个通过免疫过程与 m6A 甲基化相互作用的中心基因,并且能够有效地分类 IMS。一些研究发现 HNRNPC 与肿瘤进展之间存在关联, 46 , 47 , 48 以及免疫细胞浸润。 49 然而,哪些免疫细胞可以被HNRNPC调节,从而重塑肿瘤微环境仍然不清楚。
Our results suggest that HNRNPCs promote the activation of Treg cells. Immunosuppression in the TME, mediated by Tregs, is the main mechanism of tumor immune escape, which is associated with poor prognosis.
50
On the single‐cell level, we described the tumor immune microenvironment, especially the T cell subset. The rate of HNRNPC positive cells was significantly positively correlated with the proportion of epithelial cells and negatively correlated with the proportion of T cells. More importantly, the proportion of Treg cells in positive cells is higher, which represented a state of immunosuppression, consistent with our findings in the bulk RNA‐seq dataset. Observing the flow of communication signals between cells, we found that SPP1 and TGF‐β signal flows were activated in the positive group, which indicates immunosuppression in several cancers.
51
,
52
,
53
,
54
Furthermore, the epithelial cells in the positive group mainly interact with T cells through MIF‐(CD74+CXCR4) and MIF‐(CD74+CD44), which can mediate the formation of an immunosuppressive microenvironment caused by tumor hypoxia.
55
Our study indicated that HNRNPC can promote the infiltration of Tregs by coculture of PBMCs and PCa cells, which may be a potential mechanism by which HNRNPC promotes PCa progression through inhibiting T cell activity. Therefore, we suspected that HNRNPC could promote the infiltration of Tregs and T cell exhaustion to reshape the immunosuppressive microenvironment and promote the progression of PCa.
我们的结果表明,HNRNPC促进了 Treg 细胞的激活。肿瘤微环境中的免疫抑制,由 Treg 介导,是肿瘤免疫逃逸的主要机制,这与不良预后相关。 50 在单细胞水平上,我们描述了肿瘤免疫微环境,特别是 T 细胞亚群。HNRNPC阳性细胞的比例与上皮细胞的比例显著正相关,而与 T 细胞的比例显著负相关。更重要的是,阳性细胞中 Treg 细胞的比例更高,这代表了一种免疫抑制状态,与我们在大规模 RNA 测序数据集中发现的结果一致。观察细胞之间信号传递的流动,我们发现阳性组中的 SPP1 和 TGF-β信号流被激活,这表明在几种癌症中存在免疫抑制。 51 , 52 , 53 , 54 此外,阳性组中的上皮细胞主要通过 MIF‐(CD74+CXCR4)和 MIF‐(CD74+CD44)与 T 细胞相互作用,这可以介导由肿瘤缺氧引起的免疫抑制微环境的形成。 55 我们的研究表明,HNRNPC可以通过 PBMCs 和 PCa 细胞的共培养促进 Tregs 的浸润,这可能是HNRNPC通过抑制 T 细胞活性促进 PCa 进展的潜在机制。因此,我们怀疑HNRNPC可能促进 Tregs 的浸润和 T 细胞的耗竭,以重塑免疫抑制微环境并促进 PCa 的进展。
Our study has several limitations. First, our results were confirmed in both TCGA and GEO databases. Owing to the lack of sufficient in‐house data, the results were not externally verified. Therefore, it is necessary to validate these results in a multicenter cohort. In addition, our results showed that HNRNPC cells promoted Treg cell activation and suppression of effector CD8 T cells. However, the regulatory mechanism of m6A methylation needs to be further elucidated to improve precise immunotherapy of PCa.
我们的研究有几个局限性。首先,我们的结果在 TCGA 和 GEO 数据库中得到了确认。由于缺乏足够的内部数据,结果未得到外部验证。因此,有必要在多中心队列中验证这些结果。此外,我们的结果显示,HNRNPC细胞促进了 Treg 细胞的激活和效应 CD8 T 细胞的抑制。然而,m6A 甲基化的调控机制需要进一步阐明,以改善前列腺癌的精准免疫治疗。
Our findings provide new insights into the m6A‐TME interplay in PCa progression and present a potential target. We identified a “cold” immune phenotype in progressed PCa, and m6A reader HNRNPC regulating the abundance of Treg cells may be the mechanism of m6A methylation–mediated response to anti‐CTLA‐4. Multiple immunosuppressive phenotypes suggested that single checkpoint inhibitor is likely to produce drug resistance. Therefore, activation of the immune microenvironment through targeting upstream m6A regulators may be a potential therapeutic option for advanced PCa.
我们的研究为 m6A-TME 在前列腺癌进展中的相互作用提供了新的见解,并提出了一个潜在的靶点。我们在进展的前列腺癌中识别出了一种“冷”免疫表型,m6A 阅读器HNRNPC调节 Treg 细胞的丰度可能是 m6A 甲基化介导对抗 CTLA-4 反应的机制。多种免疫抑制表型表明,单一的检查点抑制剂可能会产生耐药性。因此,通过靶向上游 m6A 调节因子激活免疫微环境可能是晚期前列腺癌的一种潜在治疗选择。
ACKNOWLEGMENTS 致谢
None. 没有。
FUNDING INFORMATION 资金信息
This work was supported by the Medical Research Project of the Jiangsu Provincial Health and Family Planning Commission (No. H2018052) and Young Talents Program of the Jiangsu Cancer Hospital (No. 2017YQL‐04).
本研究得到了江苏省卫生和计划生育委员会医学研究项目(编号:H2018052)和江苏省肿瘤医院青年人才计划(编号:2017YQL‐04)的支持。
CONFLICT OF INTEREST STATEMENT
利益冲突声明
The authors declare no conflict of interest.
作者声明没有利益冲突。
ETHICS STATEMENT 伦理声明
Approval of the research protocol by an Institutional Reviewer Board: N/A.
研究方案获得机构审查委员会的批准:不适用。Informed Consent: N/A. 知情同意:不适用。
Registry and the Registration No. of the study/trial: N/A.
研究/试验的注册和注册号:不适用。Animal Studies: N/A. 动物研究:不适用。
Supporting information 支持信息
Supporting information S1.
支持信息 S1。
Cheng Y, Li L, Wei X, et al.
HNRNPC suppresses tumor immune microenvironment by activating Treg cells promoting the progression of prostate cancer. Cancer Sci. 2023;114:1830‐1845. doi: 10.1111/cas.15745
程 Y, 李 L, 魏 X, 等. HNRNPC 通过激活 Treg 细胞抑制肿瘤免疫微环境,促进前列腺癌的进展. 癌症科学. 2023;114:1830‐1845. doi: 10.1111/cas.15745
Yifei Cheng, Lu Li, and Xiyi Wei contributed equally to this article.
程怡菲、李璐和韦熙怡对本文贡献相同。
Contributor Information 贡献者信息
Feng Qi, Email: qf199408@163.com.
冯琦,电子邮件:qf199408@163.com。
Yanyan Zhang, Email: dabaoyan@njmu.edu.cn.
张艳艳,电子邮件:dabaoyan@njmu.edu.cn。
Xiao Li, Email: leex91@163.com.
小李,电子邮件:leex91@163.com。
DATA AVAILABILITY STATEMENT
数据可用性声明
TCGA data are openly available at https://portal.gdc.cancer.gov/.
TCGA 数据可以在 https://portal.gdc.cancer.gov/ 上公开获取。The GEO data are openly available at https://www.ncbi.nlm.nih.gov/gds.
GEO 数据可以在 https://www.ncbi.nlm.nih.gov/gds 上公开获取。
REFERENCES 参考文献
- 1. Siegel R, Miller K, Fuchs H, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71(1):7‐33. doi: 10.3322/caac.21654 [DOI] [PubMed] [Google Scholar]
- 2. Bray F, Ferlay J, Soerjomataram I, Siegel R, Torre L, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394‐424. doi: 10.3322/caac.21492 [DOI] [PubMed] [Google Scholar]
- 3. Siegel R, Miller K, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019;69(1):7‐34. doi: 10.3322/caac.21551 [DOI] [PubMed] [Google Scholar]
- 4. Halabi S, Kelly W, Ma H, et al. Meta‐analysis evaluating the impact of site of metastasis on overall survival in men with castration‐resistant prostate cancer. J Clin Oncol. 2016;34(14):1652‐1659. doi: 10.1200/jco.2015.65.7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huinen Z, Huijbers E, van Beijnum J, Nowak‐Sliwinska P, Griffioen A. Anti‐angiogenic agents – overcoming tumour endothelial cell anergy and improving immunotherapy outcomes. Nat Rev Clin Oncol. 2021;18:527‐540. doi: 10.1038/s41571-021-00496-y [DOI] [PubMed] [Google Scholar]
- 6. Yap T, Parkes E, Peng W, Moyers J, Curran M, Tawbi H. Development of immunotherapy combination strategies in cancer. Cancer Discov. 2021;11:1368‐1397. doi: 10.1158/2159-8290.Cd-20-1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beer T, Kwon E, Drake C, et al. Randomized, double‐blind, phase III trial of Ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy‐naive castration‐resistant prostate cancer. J Clin Oncol. 2017;35(1):40‐47. doi: 10.1200/jco.2016.69.1584 [DOI] [PubMed] [Google Scholar]
- 8. Maia M, Hansen A. A comprehensive review of immunotherapies in prostate cancer. Crit Rev Oncol Hematol. 2017;113:292‐303. doi: 10.1016/j.critrevonc.2017.02.026 [DOI] [PubMed] [Google Scholar]
- 9. Graff J, Alumkal J, Drake C, et al. Early evidence of anti‐PD‐1 activity in enzalutamide‐resistant prostate cancer. Oncotarget. 2016;7(33):52810‐52817. doi: 10.18632/oncotarget.10547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kwon E, Drake C, Scher H, et al. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration‐resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184‐043): a multicentre, randomised, double‐blind, phase 3 trial. Lancet Oncol. 2014;15(7):700‐712. doi: 10.1016/s1470-2045(14)70189-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gubin M, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature. 2014;515(7528):577‐581. doi: 10.1038/nature13988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang G, Zhao D, Spring D, DePinho R. Genetics and biology of prostate cancer. Genes Dev. 2018;32:1105‐1140. doi: 10.1101/gad.315739.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du K, Zhang L, Lee T, Sun T. mA RNA methylation controls neural development and is involved in human diseases. Mol Neurobiol. 2019;56(3):1596‐1606. doi: 10.1007/s12035-018-1138-1 [DOI] [PubMed] [Google Scholar]
- 14. Wang S, Sun C, Li J, et al. Roles of RNA methylation by means of N‐methyladenosine (mA) in human cancers. Cancer Lett. 2017;408:112‐120. doi: 10.1016/j.canlet.2017.08.030 [DOI] [PubMed] [Google Scholar]
- 15. Geula S, Moshitch‐Moshkovitz S, Dominissini D, et al. Stem Cells. m6A mRNA Methylation Facilitates Resolution of naïve Pluripotency toward Differentiation. Science (New York, NY). 2015;347(6225):1002‐1006. doi: 10.1126/science.1261417 [DOI] [PubMed] [Google Scholar]
- 16. Li H, Tong J, Zhu S, et al. mA mRNA methylation controls T cell homeostasis by targeting the IL‐7/STAT5/SOCS pathways. Nature. 2017;548(7667):338‐342. doi: 10.1038/nature23450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen T, Hao Y, Zhang Y, et al. M(6)a RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16(3):289‐301. doi: 10.1016/j.stem.2015.01.016 [DOI] [PubMed] [Google Scholar]
- 18. Cui Q, Shi H, Ye P, et al. mA RNA methylation regulates the self‐renewal and tumorigenesis of glioblastoma stem cells. Cell Rep. 2017;18(11):2622‐2634. doi: 10.1016/j.celrep.2017.02.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yang Y, Hsu P, Chen Y, Yang Y. Dynamic transcriptomic mA decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res. 2018;28(6):616‐624. doi: 10.1038/s41422-018-0040-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thorsson V, Gibbs DL, Brown SD, et al. The immune landscape of cancer. Immunity. 2018;48(4):812‐830 e14. doi: 10.1016/j.immuni.2018.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gibney G, Weiner L, Atkins M. Predictive biomarkers for checkpoint inhibitor‐based immunotherapy. Lancet Oncol. 2016;17(12):e542‐e551. doi: 10.1016/s1470-2045(16)30406-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Koyama S, Akbay E, Li Y, et al. Adaptive resistance to therapeutic PD‐1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. doi: 10.1038/ncomms10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cha E, Klinger M, Hou Y, et al. Improved survival with T cell clonotype stability after anti‐CTLA‐4 treatment in cancer patients. Sci Transl Med. 2014;6(238):238ra70. doi: 10.1126/scitranslmed.3008211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chow M, Ozga A, Servis R, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti‐PD‐1 therapy. Immunity. 2019;50(6):1498‐1512.e5. doi: 10.1016/j.immuni.2019.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Huang L, Malu S, McKenzie J, et al. The RNA‐binding protein MEX3B mediates resistance to cancer immunotherapy by downregulating HLA‐A expression. Clin Cancer Res. 2018;24(14):3366‐3376. doi: 10.1158/1078-0432.Ccr-17-2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chen L, Diao L, Yang Y, et al. CD38‐mediated immunosuppression as a mechanism of tumor cell escape from PD‐1/PD‐L1 blockade. Cancer Discov. 2018;8(9):1156‐1175. doi: 10.1158/2159-8290.Cd-17-1033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Aran D, Looney AP, Liu L, et al. Reference‐based analysis of lung single‐cell sequencing reveals a transitional profibrotic macrophage. Nat Immunol. 2019;20(2):163‐172. doi: 10.1038/s41590-018-0276-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Andreatta M, Corria‐Osorio J, Muller S, Cubas R, Coukos G, Carmona SJ. Interpretation of T cell states from single‐cell transcriptomics data using reference atlases. Nat Commun. 2021;12(1):2965. doi: 10.1038/s41467-021-23324-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin S, Guerrero‐Juarez CF, Zhang L, et al. Inference and analysis of cell‐cell communication using CellChat. Nat Commun. 2021;12(1):1088. doi: 10.1038/s41467-021-21246-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Giganti G, Atif M, Mohseni Y, et al. Treg cell therapy: how cell heterogeneity can make the difference. Eur J Immunol. 2021;51(1):39‐55. doi: 10.1002/eji.201948131 [DOI] [PubMed] [Google Scholar]
- 31. Bulten W, Kartasalo K, Chen P, et al. Artificial intelligence for diagnosis and Gleason grading of prostate cancer: the PANDA challenge. Nat Med. 2022;28:154‐163. doi: 10.1038/s41591-021-01620-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Scelo G, Larose TL. Epidemiology and risk factors for kidney cancer. J Clin Oncol. 2018;36:3581. doi: 10.1200/jco.2018.79.1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y, Pan C, Wang X, et al. Silencing of METTL3 effectively hinders invasion and metastasis of prostate cancer cells. Theranostics. 2021;11(16):7640‐7657. doi: 10.7150/thno.61178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Li J, Xie H, Ying Y, et al. YTHDF2 mediates the mRNA degradation of the tumor suppressors to induce AKT phosphorylation in N6‐methyladenosine‐dependent way in prostate cancer. Mol Cancer. 2020;19(1):152. doi: 10.1186/s12943-020-01267-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Z, Zhong J, Zeng J, et al. Characterization of the m6A‐associated tumor immune microenvironment in prostate cancer to aid immunotherapy. Front Immunol. 2021;12:735170. doi: 10.3389/fimmu.2021.735170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhao Y, Sun H, Zheng J, Shao C. Analysis of RNA m(6)a methylation regulators and tumour immune cell infiltration characterization in prostate cancer. Artif Cells Nanomed Biotechnol. 2021;49(1):407‐435. doi: 10.1080/21691401.2021.1912759 [DOI] [PubMed] [Google Scholar]
- 37. Zhang X, Song L, Shen J, et al. Prognostic and predictive values of immune infiltrate in patients with head and neck squamous cell carcinoma. Hum Pathol. 2018;82:104‐112. doi: 10.1016/j.humpath.2018.07.012 [DOI] [PubMed] [Google Scholar]
- 38. Knijnenburg T, Wang L, Zimmermann M, et al. Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep. 2018;23(1):239‐254.e6. doi: 10.1016/j.celrep.2018.03.076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vitale I, Shema E, Loi S, Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat Med. 2021;27(2):212‐224. doi: 10.1038/s41591-021-01233-9 [DOI] [PubMed] [Google Scholar]
- 40. Wolf Y, Bartok O, Patkar S, et al. UVB‐induced tumor heterogeneity diminishes immune response in melanoma. Cell. 2019;179(1):219‐235.e21. doi: 10.1016/j.cell.2019.08.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen Y, Chen G, Li J, et al. Association of Tumor Protein p53 and ataxia‐telangiectasia mutated Comutation with response to immune checkpoint inhibitors and mortality in patients with non‐small cell lung cancer. JAMA Netw Open. 2019;2(9):e1911895. doi: 10.1001/jamanetworkopen.2019.11895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Litchfield K, Reading J, Puttick C, et al. Meta‐analysis of tumor‐ and T cell‐intrinsic mechanisms of sensitization to checkpoint inhibition. Cell. 2021;184(3):596‐614.e14. doi: 10.1016/j.cell.2021.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bao X, Zhang Y, Li H, et al. RM2Target: a comprehensive database for targets of writers, erasers and readers of RNA modifications. Nucleic Acids Res. 2023;51(D1):D269‐D279. doi: 10.1093/nar/gkac945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Xuan JJ, Sun WJ, Lin PH, et al. RMBase v2.0: deciphering the map of RNA modifications from epitranscriptome sequencing data. Nucleic Acids Res. 2018;46(D1):D327‐D334. doi: 10.1093/nar/gkx934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Echterdiek F, Janikovits J, Staffa L, et al. Low density of FOXP3‐positive T cells in normal colonic mucosa is related to the presence of beta2‐microglobulin mutations in lynch syndrome‐associated colorectal cancer. Onco Targets Ther. 2016;5(2):e1075692. doi: 10.1080/2162402X.2015.1075692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu Z, Chen Q, Shu L, Zhang C, Liu W, Wang P. Expression profiles of m6A RNA methylation regulators, PD‐L1 and immune infiltrates in gastric cancer. Front Oncol. 2022;12:970367. doi: 10.3389/fonc.2022.970367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Guo W, Tan F, Huai Q, et al. Comprehensive analysis of PD‐L1 expression, immune infiltrates, and m6A RNA methylation regulators in esophageal squamous cell carcinoma. Front Immunol. 2021;12:669750. doi: 10.3389/fimmu.2021.669750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gruber A, Schmidt R, Ghosh S, et al. Discovery of physiological and cancer‐related regulators of 3' UTR processing with KAPAC. Genome Biol. 2018;19(1):44. doi: 10.1186/s13059-018-1415-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang S, Xu G, Chao F, Zhang C, Han D, Chen G. HNRNPC promotes proliferation, metastasis and predicts prognosis in prostate cancer. Cancer Manag Res. 2021;13:7263‐7276. doi: 10.2147/CMAR.S330713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jacobs J, Nierkens S, Figdor C, de Vries I, Adema G. Regulatory T cells in melanoma: the final hurdle towards effective immunotherapy? Lancet Oncol. 2012;13(1):e32‐e42. doi: 10.1016/s1470-2045(11)70155-3 [DOI] [PubMed] [Google Scholar]
- 51. Watermann C, Pasternack H, Idel C, et al. Recurrent HNSCC Harbor an immunosuppressive tumor immune microenvironment suggesting successful tumor immune evasion. Clin Cancer Res. 2021;27(2):632‐644. doi: 10.1158/1078-0432.CCR-20-0197 [DOI] [PubMed] [Google Scholar]
- 52. Ciummo SL, D'Antonio L, Sorrentino C, et al. The C‐X‐C motif chemokine ligand 1 sustains breast cancer stem cell self‐renewal and promotes tumor progression and immune escape programs. Front Cell Dev Biol. 2021;9:689286. doi: 10.3389/fcell.2021.689286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zeng F, Zhang Y, Han X, Zeng M, Gao Y, Weng J. Predicting non‐alcoholic fatty liver disease progression and immune deregulations by specific gene expression patterns. Front Immunol. 2020;11:609900. doi: 10.3389/fimmu.2020.609900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Pawlik A, Anisiewicz A, Filip‐Psurska B, et al. Calcitriol and its analogs establish the immunosuppressive microenvironment that drives metastasis in 4T1 mouse mammary gland cancer. Int J Mol Sci. 2018;19(7):2116. doi: 10.3390/ijms19072116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wang Y, Wang Z, Jia F, et al. CXCR4‐guided liposomes regulating hypoxic and immunosuppressive microenvironment for sorafenib‐resistant tumor treatment. Bioact Mater. 2022;17:147‐161. doi: 10.1016/j.bioactmat.2022.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information S1.
Data Availability Statement
TCGA data are openly available at https://portal.gdc.cancer.gov/.
The GEO data are openly available at https://www.ncbi.nlm.nih.gov/gds.