
Abstract 抽象
Prime editors are restricted to performing precise edits downstream of cleavage sites, thereby limiting their editing scope. Therefore, we develop inverse prime editors (iPEs) that act upstream of the nickase cleavage site by replacing nCas9-H840A with nCas9-D10A, but the editing efficiencies are limited. To address this limitation, we develop circular RNA-mediated iPEs (ciPEs), achieving editing efficiencies ranging from 0.1% to 24.7%. Further optimization using Rep-X helicase increases editing efficiencies to a range of 2.7%–55.4%. The Rep-X-assisted ciPE system thus expands the scope of editing and improves efficiencies at genomic sites that are previously difficult to target. The Rep-X-assisted ciPE system will complement canonical PE system in enabling more extensive and efficient editing across a wider range of the human genome.
Prime 编辑者只能在切割位点的下游进行精确编辑,从而限制了他们的编辑范围。因此,我们开发了反向引物编辑器 (iPE),它通过用 nCas9-D10A 取代 nCas9-H840A 起作用在切口酶切割位点的上游,但编辑效率是有限的。为了解决这一限制,我们开发了环状 RNA 介导的 iPE (ciPE),实现了 0.1% 至 24.7% 的编辑效率。使用 Rep-X 解旋酶进行进一步优化可将编辑效率提高到 2.7%–55.4% 的范围内。因此,Rep-X 辅助 ciPE 系统扩大了编辑范围,并提高了以前难以靶向的基因组位点的效率。Rep-X 辅助 ciPE 系统将补充经典 PE 系统,在更广泛的人类基因组范围内实现更广泛、更高效的编辑。
Similar content being viewed by others
其他人正在查看类似内容


Introduction 介绍
Precise genome editing is essential in various fields such as biological research, medical studies, and crop breeding1,2,3,4. Prime editors, which generate precise base substitutions and accurate insertions and deletions of small DNA fragments, are powerful tools for achieving such precision5,6,7,8. Canonical prime editing system utilizes a reverse transcriptase together with a non-target strand (NTS) nickase, such as nCas9-H840A9,10,11 or nCas12a-R1138A12, to produce edits downstream of the cleavage sites (Fig. 1a). However, due to the absence of reverse transcriptases capable of polymerizing DNA in the 3′ → 5′ direction13,14, prime editors are generally limited to producing precise edits downstream of their cleavage sites (Fig. 1a), thereby limiting the editing scope. In this study, we report the development of an inverse prime editor (iPE) system utilizing a target strand (TS) nickase, nCas9-D10A, for inverse prime editing. We initially develop iPE editors by replacing the nCas9-H840A nickase with nCas9-D10A. However, these iPE editors exhibit limited prime editing efficiency, with a maximum editing rate of only 8.6%. We hypothesize that this limitation might be due to inefficient unwinding of the primer binding site (PBS) within the iPE editors. To address this issue, we design circular RNA-mediated iPEs (ciPEs) that utilize the unique properties of circular RNA to enhance the efficiency of inverse prime editing. The ciPE editors result in a significant improvement, with an efficiency of 0.1%–24.7%. To further improve efficiency, we incorporate a modified 3′ → 5′ helicase, Rep-X, as an auxiliary protein, and this increases inverse editing efficiency to 2.7%–55.4%. Our Rep-X-assisted, circular RNA-mediated ciPE system widens the scope of prime editing and improves efficiency at genomic sites that are previously difficult to target.
精确的基因组编辑在生物学研究、医学研究和作物育种等各个领域都是必不可少的 1,2,3,4。Prime 编辑器可生成精确的碱基替换以及小 DNA 片段的准确插入和缺失,是实现这种精确度的强大工具 5,6,7,8。经典引物编辑系统利用逆转录酶和非靶链 (NTS) 切口酶(如 nCas9-H840A 9,10,11 或 nCas12a-R1138A12)在切割位点下游产生编辑(图 1a)。然而,由于缺乏能够在 3' → 5' 方向聚合 DNA 的逆转录酶 13,14,因此引物编辑器通常仅限于在其切割位点下游产生精确的编辑(图 1a),从而限制了编辑范围。在这项研究中,我们报告了利用靶链 (TS) 切口酶 nCas9-D10A 进行反向引物编辑的反向引物编辑器 (iPE) 系统的开发 。我们最初通过用 nCas9-D10A 替换 nCas9-H840A 切口酶来开发 iPE 编辑器。然而,这些 iPE 编辑器表现出有限的 prime 编辑效率,最大编辑率仅为 8.6%。我们假设这种限制可能是由于 iPE 编辑器内引物结合位点 (PBS) 的解旋效率低下。为了解决这个问题,我们设计了环状 RNA 介导的 iPE (ciPE),它利用环状 RNA 的独特特性来提高反向引物编辑的效率。 ciPE 编辑器带来了显著的改进,效率为 0.1%–24.7%。为了进一步提高效率,我们加入了修饰的 3′ → 5′ 解旋酶 Rep-X 作为辅助蛋白,这将反向编辑效率提高到 2.7%–55.4%。我们的 Rep-X 辅助环状 RNA 介导的 ciPE 系统拓宽了引物编辑的范围,并提高了以前难以靶向的基因组位点的效率。
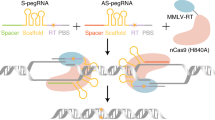
图 1:用于人类细胞反向引物编辑的 iPE 的开发。

a Schematic diagram of the canonical prime editor (PE) using nCas9-H840A. The red region of the genome represents the DNA segment that can be edited by PE. b Schematic diagram of the inverse prime editor (iPE) using nCas9-D10A. The red region of the genome represents the DNA segment that can be edited by iPE. c Inverse prime editing frequencies achieved by four different iPE constructs, including iPE2–5, at target sites HBB, HEXA, FANCF, and PDCD1 in HEK293T cells. d Inverse prime editing frequencies achieved by eight different iPE constructs, including iPE2–5 and iPEmax2–5, at target sites DMD, ETS1-T1, FANCF, and PAH in HEK293T cells. Frequencies (mean ± s.e.m.) in c-d were obtained from three biological replicates (n = 3). Ins, insertion; Del, deletion. InDels, byproducts of random insertions and deletions. Source data are provided as a Source Data file.
Results
Developing inverse prime editors (iPEs) using nCas9-D10A
A prime editor generally contains three components: a Cas9 nickase (nCas9-H840A), a reverse transcriptase (Moloney-murine leukemia virus reverse transcriptase, M-MLV RT), and a prime editing guide RNA (pegRNA) (Fig. 1a)5,7. To achieve prime editing upstream of the cleavage site (inverse prime editing), we utilized nCas9-D10A nickase15 instead of nCas9-H840A to generate inverse prime editors (iPE editors, Fig. 1b). The nCas9-D10A nickase has been demonstrated to exhibit more specific nickase activity, resulting in fewer double-strand breaks (DSBs) and producing fewer insertion and deletion (InDel) byproducts15. Initially, we developed iPE2 utilizing the nCas9-D10A, M-MLV RT, and pegRNA. Additionally, we developed three other inverse prime editors: iPE3, by adding a nicking sgRNA5 based on iPE2; iPE4, by adding a negative inhibitor of MLH1 (MLH1dn)9; and iPE5, by integrating both factors, based on the corresponding canonical prime editors PE2, PE3, PE4, and PE5.
We initially tested the inverse prime editing efficiencies of iPE editors, including iPE2, iPE3, iPE4, and iPE5, at HBB, HEXA, FANCF, and PDCD1 target sites in HEK293T cells (Fig. 1c). We found that these iPE editors produced only very low prime editing efficiencies, ranging from 0.04 to 1.06% at four target sites, and the average editing efficiency was 0.3%. The PEmax editors with non-target DNA strand nickase, H840A-Cas9, have been shown to perform more efficiently than PE editors9. We constructed iPEmax editors using target DNA strand nickase, D10A-Cas9, instead of non-target DNA strand nickase of H840A-Cas9, and inverse pegRNAs based on PEmax, including iPEmax2, iPEmax3, iPEmax4, and iPEmax5, to perform inverse prime editing. We tested the inverse prime editing efficiencies of iPEmax editors at DMD, ETS1-T1, FANCF, and PAH target sites in HEK293T cells (Fig. 1d). The results showed that the iPEmax editors still produced low inverse prime editing efficiencies (average 2.0% for iPEmax2–5 at the four sites, maximum 8.6% for iPEmax2 with epegRNA at the DMD site), although the inverse editing efficiencies of the iPEmaxs were higher than those of the iPE editors (Fig. 1d).
Current studies suggest that the high efficiency of canonical prime editors is largely due to the placement of their primer binding sites (PBSs) within the R-loop region produced by the nickase16,17. CRISPR-Cas9 can efficiently unwind DNA double strands within a 20-bp range16,17, facilitating PBS binding and enabling M-MLV RT to generate flap sequences containing the desired edits, thereby achieving efficient prime editing through intracellular repair systems5,6,7,8. However, in the inverse prime editors, the PBS downstream of the target site where DNA unwinding is less efficient, thereby significantly hindering reverse transcription and leading to reduced efficiency of inverse prime editing (Fig. 1c, d).
We therefore sought to develop an efficient method for achieving inverse prime editing at target sites. Previous studies have suggested that cellular repair systems temporarily unwind the double helix near double-strand breaks (DSBs) during the repair process18,19. In addition, it has been shown that prime editors developed with WTCas9 (wild-type Cas9) exhibit greater efficiency than canonical prime editors20,21. We hypothesized that DSBs induced by WTCas9 might activate repair systems to unwind the DNA, thereby facilitating PBS binding to single-strand DNA, thereby enhancing inverse prime editing efficiency. Therefore, we developed nuclease-dependent inverse prime editors (nu-iPEs) with WTCas9 (Supplementary Fig. 1a). We tested the nu-iPE2 in a HEK293T GFP reporter system. This GFP reporter system contains a one-base “G” deletion and two substitutions of “C” and “G” in the GFP coding sequences, which result in a frameshift and amino acid substitutions, respectively. Only precise inverse prime editing events can restore the fluorescence of GFP (Supplementary Fig. 1b, c). We observed that the inverse prime editing efficiency of nu-iPE2 was significantly higher than that of the nickase-dependent inverse prime editor iPE2 (Supplementary Fig. 1d). This increased efficiency was also observed at endogenous target sites in RNF2, RUNX1-T1, DMD, and FANCF in HEK293T cells, where nu-iPE2 achieved inverse prime editing frequencies of 1.8%, 3.1%, 3.4%, and 5.8%, respectively, representing 6.2-, 35.9-, 1.7-, and 32.1-fold improvements over iPE2 (Supplementary Fig. 1e).
Circular RNA-mediated inverse prime editors using nCas9-D10A
Although nuclease-dependent inverse prime editors demonstrated more efficient inverse prime editing, they also generated many InDel byproducts, which limit their potential for treating genetic diseases1,5. To address this, we sought another method to improve the efficiency of inverse prime editing without inducing extensive DSBs, thereby reducing the occurrence of InDel byproducts. We focused on circular RNA to improve editing efficiency. Previous studies have shown that circular RNAs can bind to specific genomic regions, generating R-loop structures that regulate gene expression and influence various biological functions of genes22,23,24,25. Moreover, the loop structure of circular RNAs makes them resistant to degradation by RNA exonucleases, leading to a more stable state of the RTT-PBS components of pegRNA26, which has been shown to be important for achieving high prime editing efficiency27,28.
We hypothesized that circular RNA might aid in unwinding DNA upon stable binding, thereby enhancing inverse prime editing. To test this, we developed nickase-dependent circular RNA-mediated inverse prime editors (ciPE editors), consisting of the nCas9-D10A (with only the D10A substitution relative to wild type), MCP-M-MLV RTΔRNase H (with the RNase H domain removed from the C terminal M-MLV RT reverse transcriptase)12, as well as an sgRNA, and a circular RNA containing reverse transcriptase template (RTT) and PBS sequences, using the “Tornado” circular RNA durable expression system29 (Fig. 2a, b). The circular RNA also contained two MS2 hairpin sequences for recruiting MCP-M-MLV RTΔRNase H. We tested these inverse prime editing editors at various target sites in HEK293T cells (Fig. 2c). Notably, the editing efficiency of circular RNA-mediated ciPE2 was significantly higher than that of split iPE2, which uses M-MLV RTΔRNase H. Specifically, the inverse editing efficiency of circular RNA-mediated ciPE2 at the DMD, FANCF, HEK3, and RNF2 sites reached 7.5%, 2.0%, 0.7%, and 1.5% (average 2.9%), respectively, showing 3.6-, 11.1-, 6.1-, and 11.8-fold efficiency increases compared to split iPE2 (Fig. 2c). We also found that a control treatment with ciPE2 editor using nCas9-H840A produced a low number of inverse prime edits (up to 0.39% at the DMD site) (Fig. 2c). We speculate that this may be due to nCas9-H840A’s residual ability to cut the target DNA strand, consistent with previous studies15.

a Schematic diagram of circular RNA-mediated inverse prime editors (ciPEs). b Schematic diagrams of the structure of ciPE editors. CMV, the CMV promoter of cytomegalovirus; U6, the polymerase III promoter of U6; NLS, bipartite nuclear localization signal; M-MLV RTΔRNase H, deletion variant of M-MLV RT with no RNase H domain; 5′ RL, 5′ ribozyme and ligation sequences; 3′ RL, 3′ ribozyme and ligation sequences; RTT, reverse transcriptase template; PBS, primer binding site. c Comparison of inverse prime editing frequencies between split iPE2 and ciPE2 at four target sites in HEK293T cells. Frequencies (mean ± s.e.m.) in c were obtained from three biological replicates (n = 3). d Comparison of inverse prime editing efficiencies between ciPE2–5 and split iPEmax2–5 using epegRNA at six target sites in HEK293T cells. Frequencies (mean ± s.e.m.) in d were obtained from four biological replicates (n = 4). circRNA, circular RNA; Ins, insertion; Del, deletion. InDels, byproducts of random insertions and deletions. P values were obtained from two-tailed Student’s t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are provided as a Source Data file.
To confirm the efficiency of other ciPE editors, we developed three additional variants: ciPE3, by adding a nicking sgRNA producing a nick in the non-target strand (NTS) to ciPE25; ciPE4, by adding a negative inhibitor of MLH1 (MLH1dn)9; and ni-ciPE5, by including both factors. We then compared the inverse prime editing of these ciPE editors to that of epegRNA-mediated split iPEmax editors at the PDCD1, CXCR4, HEXA, HEK4, BCL11A, and TRAC target sites (Fig. 2d). The results showed that ciPE editors had inverse prime editing efficiencies ranging from 0.1% to 24.7% (average 5.2%), while epegRNA-mediated split iPEmax editors reached only from 0.0% to 11.0% (average 2.6%) editing efficiencies across all six sites. At the PDCD1, CXCR4, HEXA, HEK4, and BCL11A sites, the ciPE editors had average editing efficiencies of 4.7%, 0.8%, 9.8%, 13.4%, and 2.1%, respectively, which were 4.8-, 2.1-, 2.0-, 1.7, and 1.4-fold greater, respectively, than those generated by epegRNA-mediated split iPEmax editors (Fig. 2d). The epegRNA-mediated split iPEmax editors generated very few edits (<0.01%) at the TRAC target site, while the ciPE editors had significantly higher editing efficiencies, reaching 1.8%, with an average efficiency 1131.0-fold greater than those of the epegRNA-mediated split iPEmax editors (Fig. 2d).
To further enhance inverse prime editing, we tested nuclease-dependent circular RNA-mediated inverse prime editors (nu-ciPE editors) that incorporated WTCas9 and circular RNAs (Supplementary Fig. 2a, b). We compared the editing efficiencies of nu-ciPE editors with nu-iPEmax editors at six endogenous target sites in HEK293T cells and found that nu-ciPE2 and nu-ciPE4 significantly increased inverse prime editing efficiencies (1.2–76.2-fold) compared to nu-iPEmax2 and nu-iPEmax4 at all six target sites, with nu-ciPE2 reaching an editing frequency of 18.9% at the PAH site (Supplementary Fig. 2c).
Developing helicase Rep-X-assisted ciPEs
These results indicated that the effectiveness of DNA unwinding can further improve inverse prime editing. Considering this, and to avoid the DSBs induced by WTCas9, we explored the use of DNA helicases. DNA helicases are enzymes that unwind DNA double strands by hydrolyzing ATP to break hydrogen bonds between nucleotides30,31,32. DNA helicases can be divided into two classes based on their movement direction: those that unwind DNA in the 5′ → 3′ direction and those in the 3′ → 5′ direction31. In inverse prime editors, nCas9-D10A generates an R-loop structure at the target site, forming a single-stranded DNA of approximately 20-bp around the target site and cleaving the target strand at the 17-bp position to produce a small single DNA strand at the 3′ terminal. Subsequently, we hypothesize that helicases may bind to this small single-stranded region, continue to unwind the DNA double strand, and create a longer single-stranded region, facilitating PBS binding, thus enhancing reverse transcription and potentially increasing the efficiency of inverse prime editing.
We therefore selected a modified 3′ → 5′ helicase Rep-X from Escherichia coli31 and constructed Rep-X-assisted circular RNA-mediated inverse prime editing editors (Fig. 3a, b) to increase the efficiency of inverse prime editing. We further tested the efficiencies at six additional sites to confirm the effect of Rep-X on ciPE2. At the HEK4, DMD, BCL11A, PSMB2, GFAP, and HEXA target sites, Rep-X-assisted ciPE2 editor (average 9.1%) achieved more efficient inverse editing than ciPE2 editor (average 6.6%) alone, reaching up to 14.6% efficiency at the DMD site in HEK293T cells (Fig. 3c). We further constructed Rep-X-assisted ciPE editors, including ciPE3, ciPE4, and ciPE5, and tested them at the MECP2 site. The results showed that Rep-X-assisted ciPEs performed on average 4.6-fold more efficient precise editing than ciPEs (Supplementary Fig. 3a). We also found Rep-X-assisted nu-ciPE editors performed an average of 17.0% and 23.0% inverse prime editing, 1.7- and 1.4-fold more efficient precise editing than nu-ciPEs at the DMD and RUNX1-T1 sites (Supplementary Fig. 3b). The Rep-X-assisted ciPE editors performed efficient inverse prime editing, with efficiencies ranging from 7.4% to 14.1% at the GFAP site, 11.1% to 55.4% at the HEK4 site, and 10.7% to 16.4% at the DMD site in HEK293T cells (Fig. 3d). The average editing efficiencies of ciPE editors and Rep-X-assisted ciPE editors were 11.4% and 17.0%, respectively, at the three sites (Fig. 3d). We constructed ciPEmax editors by combining MCP, D10A-Cas9, and M-MLV RTΔRNase H based on PEmax, as well as the ciPE7 editors by further incorporating the small La protein33. However, these editors yielded lower inverse prime editing efficiencies compared to the ciPE editors (Supplementary Fig. 4). We also examined additional single-base and >3-bp editing events using the Rep-X-assisted ciPE editors. The 1-bp substitution, 6-bp substitution, and 6-bp deletion at the DMD site, and 5-bp deletion, 10-bp substitution, and 1-bp substitution at the GFAP site yielded inverse prime editing efficiencies of 16.3%, 15.1%, 13.5%, 10.8%, 7.3%, and 3.5%, respectively (Fig. 3e). In addition, we determined numbers of Rep-X-assisted ciPE edits in cells of different cell lines, and the frequencies of Rep-X-assisted ciPE4 and ciPE5 edits in HeLa, K562, and U2OS cells averaged 9.1%, 10.2%, and 11.7%, respectively, at the HEK4 site, and 17.7%, 18.3%, and 19.4% at the DMD site (Fig. 3f). Potential cytotoxic effects of ciPE and Rep-X-assisted ciPE systems on the viability of HEK293T cells were also examined, and we found that these systems were no more cytotoxic than the canonical PE system (Supplementary Fig. 5). Circular RNA-mediated inverse prime editors thus exhibited significantly higher inverse prime editing efficiencies when assisted by the Rep-X helicase, with editing efficiency reaching 55.4%.

a Schematic diagram of Rep-X-assisted ciPE. Rep-X is included to aid in unwinding DNA to enhance editing efficiency. b Schematic diagrams of structure of Rep-X-assisted ciPE editors. For abbreviations, see Fig. 2b. c Comparison of inverse prime editing efficiencies between Rep-X-assisted ciPE2 and ciPE2 at six target sites in HEK293T cells. Frequencies (mean ± s.e.m.) at HEK4, DMD, BCL11A, PSMB2, and GFAP sites were obtained from eight biological replicates (n = 8) and three biological replicates (n = 3) at HEXA site. d Comparison of inverse prime editing efficiencies between three ciPE constructs and three Rep-X-assisted ciPE constructs at the GFAP, HEK4, and DMD target sites in HEK293T cells. Frequencies (mean ± s.e.m.) were obtained from eight biological replicates (n = 8). e Inverse prime editing efficiencies at the DMD and GFAP sites in HEK293T cells using Rep-X-assisted ciPE4 and ciPE5 editors. Frequencies (mean ± s.e.m.) were obtained from three biological replicates (n = 3). f Inverse prime editing efficiencies at the HEK4 and DMD sites in HeLa, K562, and U2OS cells using Rep-X-assisted ciPE4 and ciPE5 editors. Frequencies (mean ± s.e.m.) were obtained from four biological replicates (n = 4). Del, deletion. InDels, byproducts of random insertions and deletions. P values were obtained from two-tailed Student’s t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are provided as a Source Data file.
Comparing Rep-X-assisted ciPEs with existing prime editing systems
We also compared Rep-X-assisted ciPEs with existing prime editing systems. Previous studies had shown that PAM-flexible Cas9 variants would be used to develop prime editors34. Accordingly, we constructed PAMless PEs based on the strategy of prime editors with PAM flexibility including SpRY and SpG34 (Fig. 4a). We then compared the PAMless PEs and Rep-X-assisted ciPEs at DMD, GFAP, P2RY1 (Fig. 4b), GFAP-T2, and IPP sites (Supplementary Fig. 6a, b) and found that the PAMless PEs only achieved average editing efficiencies of 0.4%, 4.5%, 0.7%, 0.2%, and 0.04%, respectively, at the five targets. The Rep-X-assisted ciPEs achieved average editing efficiencies of 14.0%, 14.0%, 6.0%, 6.8%, and 5.8%, respectively, representing 33.7-, 3.1-, 8.4-, 35.4-, and 160.6-fold improvements (Fig. 4b and Supplementary Fig. 6a, b). We also compared Rep-X-assisted ciPEs with the twinPE, which has been shown to produce edits in genome regions farther from the PAM sequence8 (Supplementary Fig. 7a). The twinPE editor produced a low prime editing efficiency of only 0.8% at the PSMB2 site, while the Rep-X-assisted ciPE editors achieved much higher editing efficiencies ranging from 14.1% to 17.7%, representing an average 19.4-fold increase in efficiency (Supplementary Fig. 7b). Finally, we compared the Rep-X-assisted ciPE system with the PE7 system at genomic sites where both systems are expected to perform well (Supplementary Fig. 8a). The PE7 system achieved 4.8%, 23.9%, and 15.8% edits at the HEK4, DMD, and GFAP sites, compared with 51.4%, 16.4%, and 12.5% by the Rep-X-assisted ciPE system (Supplementary Fig. 8b). These findings indicated that Rep-X-assisted ciPE editors consistently demonstrated higher editing efficiencies than canonical PE system, twinPE system, and PAMless PE editors, especially at genomic sites that were previously difficult to target. Additionally, at sites where both Rep-X-assisted ciPE and canonical PE systems were predicted to be effective, the Rep-X-assisted ciPE system achieved substantially higher prime editing efficiency at specific genomic loci.

a Schematic diagrams of prime editing performed by Rep-X-assisted ciPEs and PAMless-PEs at the target sites. b Comparison of prime editing efficiencies at the DMD, GFAP, and P2RY1 sites in HEK293T cells using Rep-X-assisted ciPEs and PAMless-PEs. Frequencies (mean ± s.e.m.) at DMD and GFAP sites were obtained from four biological replicates (n = 4) and three biological replicates (n = 3) at the PSMB2 site. c Schematic diagrams of prime editing performed by Rep-X-assisted ciPEs, PAMless-PEs, and twinPEs at the disease sites that are difficult to target with canonical PEs. d Comparison of prime editing efficiencies at the BRCA1 and RPE65 disease sites39 in HEK293T cells using Rep-X-assisted ciPEs, PAMless-PEs, and twinPEs. Frequencies (mean ± s.e.m.) were calculated from three biological replicates (n = 3). Del, deletion; dup, duplication. InDels, byproducts of random insertions and deletions. P values were obtained from two-tailed Student’s t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Source data are provided as a Source Data file.
Prime editing of disease sites using Rep-X-assisted ciPEs
To examine the potential applications of the Rep-X-assisted ciPE system, we searched disease-associated mutations in BRCA1 and RPE65 (Fig. 4c). Deletion of bases 3756 ~ 3759 in the coding sequence of the BRCA1 gene results in a frameshift mutation after Ser1253 and can lead to breast-ovarian cancer. Similarly, duplication of the 89th base in the CDS of the RPE65 gene results in a frameshift mutation after Thr31, causing Leber congenital amaurosis. Using the Rep-X-assisted ciPE system, we mimicked these disease-causing mutations and achieved 13.3% and 9.5% inverse prime editing efficiencies at the disease sites in BRCA1 and RPE65, respectively (Fig. 4d). The PAMless PEs and twinPE systems yielded much lower editing efficiencies—5.7% and 2.9%, and 0.8% and 0.5%, respectively, at these two targets (Fig. 4d). The Rep-X-assisted ciPE system is therefore superior to the PAMless PE and twinPE editors at disease targets upstream of cleavage sites and is also able to generate models of disease.
Prime editors are known to produce few off-target edits, but they tend to produce more by products, which limits their clinical applications5,12. Since nCas9-D10A exhibits higher precision than nCas9-H840A15, we compared the precision of editing between Rep-X-assisted ciPE and canonical PE systems. We found that the canonical PE system produced editing purity of 87.9%, 88.1%, and 91.3% at the GFAP, HEK4, and DMD sites, respectively, while the Rep-X-assisted ciPE editors produced higher purity—96.6%, 92.1%, and 94.7%—at the same sites (Supplementary Fig. 9a). We also compared the editing purity of the Rep-X-assisted ciPE editors and the PAMless PE editors. The results showed that the PAMless PE editors produced 12.5% editing by products at the PSMB2 site, substantially more than the Rep-X-assisted ciPE editors (3.9%) (Supplementary Fig. 9b). We also noted that the editing purity of Rep-X-assisted ciPE editors at the disease target BRCA1 reached 96.7%, much higher than the purities achieved with the PAMless PE editor (72.3%) and twinPE editor (79.1%) (Supplementary Fig. 9c). Overall, the Rep-X-assisted ciPE system based on D10A-Cas9 and circular RNA possesses greater precision and may therefore be safer to use.
Off-target effects of circular RNA-mediated inverse prime editors
To assess the off-target effects of circular RNA-mediated inverse prime editors, endogenous off-target sites were identified using Cas-OFFinder35. We examined nine off-target sites in GFAP, nine in HEK4, and eight in DMD for editing by ciPE2, ciPE3, ciPE4, and ciPE5 (Fig. 5a), as well as by Rep-X-assisted ciPE2, ciPE3, ciPE4, and ciPE5 (Fig. 5b). At the GFAP and DMD target sites, only background levels of InDels were detected at the off-target sites for GFAP ( < 0.04%) and DMD ( < 0.01%), with no inverse prime editing observed at any off-target sites (Fig. 5a, b). Similarly, at the HEK4 site, we observed only background levels of InDels (<0.76%), with no precise inverse prime editing detected at eight of the nine off-target sites (Fig. 5a, b). However, the off-target site, off-target-3, at the HEK4 site showed 1.32%–6.32% InDels and 0.05%–0.67% inverse prime editing by ciPE editors and Rep-X-assisted ciPE editors (Fig. 5a, b). It is known that prime editors generally produce few InDel byproducts and that off-target prime edits are difficult to produce unless the sequences between on-target and off-target are very similar12. Upon closer inspection of the off-target-3 sequences, we found them to be highly similar to the on-target sequences, with only two mismatched bases at positions 6 and 7 of the unseeded sequences. In addition, seven of the eight bases in the PBS positions of ciPE editors were identical at the HEK4 site (Fig. 5a, b). Therefore, highly similar target sequences and downstream sequences contribute to the low number of InDels and off-target inverse prime edits observed. These results suggested that when using ciPE editors for inverse prime editing, it is crucial to select highly specific targets and pay attention to the similarity of downstream sequences to ensure accuracy, given that the editing and PBS regions of inverse prime editors differ from those of canonical prime editors.

a Mutation frequencies achieved using ciPEs at three on-target and 26 off-target sites identified by Cas-OFFinder for GFAP, HEK4, and DMD in HEK293T cells. b Mutation frequencies achieved using Rep-X-assisted ciPEs at three on-target and 26 off-target sites identified by Cas-OFFinder for GFAP, HEK4, and DMD in HEK293T cells. Frequencies (mean ± s.e.m.) in a, b were calculated from three biological replicates (n = 3). The sequences in the red box represent similar primer binding sequences downstream of the cleavage site between on-target and off-target at the HEK4 target site. Del, deletion. InDels, byproducts of random insertions and deletions. Source data are provided as a Source Data file.
Discussion
In summary, the Rep-X-assisted, circular RNA-mediated inverse prime editing significantly expands the editing range of prime editing, enhancing efficiency at genomic sites that were previously difficult to target. The combination of nCas9-H840A-based canonical prime editing and nCas9-D10A-based inverse prime editing offers the potential to significantly extend the target range and improve editing efficiency while reducing the generation of by products. Studies have shown that the single-strand cleavage activity of nCas9-D10A is more precise and results in fewer InDel byproducts compared to nCas9-H840A15. Consequently, our nCas9-D10A-based inverse prime editors are expected to produce fewer InDel byproducts. The use of nCas9-D10A in the prime editors may also provide inspiration for the development of more editors. For example, our study found that ciPEs performed efficient A-to-G or C-to-T base editing (Supplementary Fig. 10a, b).
Previous research has indicated that many prokaryotic genome editing systems and those that are effective in vitro exhibit less efficiency in eukaryotic cells36,37, likely due to difficulties in unwinding double-stranded DNA38. The superior gene editing capabilities of CRISPR-Cas9 are heavily dependent on its DNA unwinding ability38. Circular RNA, with its stability, durability, and low immunogenicity26, can bind to genomic DNA to form R-loops22,23,24,25, facilitating DNA unwinding, which is likely to play a crucial role in regulating gene expression and biological function. The helicase Rep-X used in this study also stimulates DNA unwinding and could be further optimized by rational design and directed evolution. We anticipate that circular RNA and helicases will become increasingly important for progress in genome editing. We also found that the ciPEmax editors constructed by concatenating MCP, nCas9-D10A, and M-MLV RTΔRNase H, as well as ciPE7 editors, which further incorporate the small La protein, exhibited lower efficiency in inverse prime editing compared to ciPE. Based on the development of previous CPE editors12, we speculate that circular RNA-mediated prime editors operate through a different mechanism compared to canonical PE editors. This suggests that current ciPE editors still have significant potential for optimization.
Methods
Plasmid construction
The sgRNA, nicking sgRNA, pegRNA, and epegRNA vectors driven by the human U6 promoter were constructed by annealing oligonucleotides and inserting them into the pOsU3 backbone (Addgene #170132) using the Golden Gate assembly method. The circular RNA expression vector backbone was synthesized using primers with sequences specified by the “Tornado” circular RNA expression system29. The RTT and PBS sequences in the circular RNA expression vectors (Supplementary Data 1) of ciPE system were introduced into the prime editor vector backbone (Supplementary Data 2) using the Golden Gate assembly method. The iPE, iPEmax, nu-iPE, nu-iPEmax, twinPE, SpG-PE, SpRY-PE, nu-ciPE, ciPE, and Rep-X-assisted ciPE vectors were derived from the pCMV-PE2 vector (Addgene #132775) using a Uniclone One Step Seamless Cloning Kit (Genesand, CN). All DNA oligonucleotides were synthesized by Beijing Genomics Institute (BGI). DNA fragments for plasmids cloning were obtained by PCR amplification using Phanta Max Master Mix (Vazyme, CN). Endonucleases were obtained from New England Biolabs (NEB, USA).
Culture of mammalian cells
HEK293T cells (American Type Culture Collection (ATCC), CRL-3216) and HeLa cells (ATCC, CRM-CCL-2) were cultured in Dulbecco’s modified Eagle’s medium (Gibco, USA) supplemented with GlutaMax. U2OS cells (ATCC, HTB-96) were cultured in McCoy’s 5A Medium Modified (Gibco, USA), and K562 cells (ATCC, CCL-243) were cultured in Iscove’s modified Dulbecco’s medium (Gibco, USA) plus GlutaMax, supplemented with 10% (v/v) fetal bovine serum (Gibco, USA), 0.1% (v/v) penicillin-streptomycin (Gibco, USA), and 5 mg/L Plasmocin Prophylactic (InvivoGen, USA). All cell lines were grown at 37 °C with 5% CO2 and subcultured for 3 days. They were tested for mycoplasma using a Mycoplasma Detection Kit (TransGen Biotech, CN).
Transfection and lysis of mammalian cells
HEK293T, HeLa, K562, and U2OS cells were seeded at a density of 16,000 cells per well in poly-D-lysine-coated 96-well clear plates (Corning, USA). After 16 to 20 h of incubation, they were transfected with 150 ng of iPE (including iPE, nu-iPE, iPEmax, and nu-iPEmax) plasmids and 50 ng each of pegRNA, epegRNA, and nicking sgRNA plasmids, using 0.4 μL of Lipofectamine 2000 (Invitrogen, USA) according to the manufacturer’s protocol. For circular RNA-mediated inverse prime editing (ciPE), HEK293T, HeLa, K562, and U2OS cells were transfected with 150 ng of ciPE or Rep-X-assisted ciPE plasmids and 50 ng each of sgRNA, circular RNA, and nicking sgRNA plasmids. The transfected cells were further cultured at 37 °C with 5% CO2.
Preparation of genomic DNA for sequencing
To prepare genomic DNA from HEK293T, HeLa, K562, and U2OS cells, the medium was removed from the wells containing cells 72 h after transfection, and the cells were lysed in 100 μL lysis buffer containing 2 μL of 25 μg/mL proteinase K and incubated at 55 °C for 30 min in 96-well plates, followed by an incubation at 95 °C for 5 min to inactivate the proteinase K. The generated genomic DNA suspensions were directly used as templates for the PCR amplification using Phanta Max Master Mix (Vazyme, CN).
Amplicon deep sequencing and data analysis
The genomic region of interest, approximately 150–200 bp (PCR Round 1), was amplified by PCR in a 20 μL reaction volume containing 10 pmol of forward and reverse primer, 10 μL of Phanta Max Master Mix (Vazyme, CN), 1 μL lysed genomic DNA and 7 μL ddH2O. PCR was performed under the following conditions: 95 °C for 3 min, followed by 34 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 20 s, with a final extension at 72 °C for 5 min. Next, in a second PCR (PCR Round 2), 1 μL product from PCR round 1 was amplified in 30 μL mixtures containing 10 pmol of unique forward and reverse Illumina primer pairs with barcodes, 15 μL Phanta Max Master Mix (Vazyme, CN), and 12 μL ddH2O, under the following conditions: 95 °C for 3 min, followed by 10–13 cycles of 95 °C for 15 s, 60 °C for 15 s, and 72 °C for 20 s, with a final extension at 72 °C for 5 min.
After confirming that all the samples were clear using agarose gel electrophoresis, the products of PCR Round 2 from each reaction were mixed, electrophoresed on a 1.2% agarose gel, purified using a GeneJET Gel Extraction Kit (Thermo Fisher Scientific, USA), and quantified with a NanoDrop 2000 (Thermo Fisher Scientific, USA). This library was normalized and sequenced on an Illumina MiSeq platform (Qi Biodesign, CN). Amplicon sequencing data was analyzed using the published code (https://github.com/ReiGao/GEanalysis). Each treatment was performed in three independent biological replicates. Primer sequences for PCR Round 1 and PCR Round 2 are shown in Supplementary Data 3 and 4, respectively.
Assessing editing efficiencies by green fluorescence
To evaluate the genome editing efficiencies of ciPEs, 40,000 HEK293T cells were transfected with 300 ng of iPE and nu-iPE plasmids and 20 ng of GFP reporter plasmids, and 100 ng of pegRNA plasmids in Poly-D-Lysine-coated 48-well clear plates (Corning, USA). After 2 days of incubation at 37 °C with 5% CO2, green fluorescence was measured using a Leica microsystem (Leica, DE).
Analysis of off-target editing
Potential off-target sites were identified using the online tool Cas-OFFinder35 (http://www.rgenome.net/cas-offinder/). The off-target sites analyzed in this study had three or fewer nucleotide mismatches compared to the on-target sites. Amplicons were sequenced using the MiSeq platform (Qi Biodesign, CN), with each off-target site examined with three independent samples. Sequence information and primers for each off-target site are listed in Supplementary Data 5 and 6.
Cell viability assays
Cell viability assays were performed using the CellTiter 96® AQueous One Solution Cell Proliferation Kit (Promega, USA), according to the manufacturer’s instructions. HEK293T cells were transfected at 80%–90% confluency. Seventy-two hours post-transfection, 20 μL of the CellTiter 96® AQueous One Solution Reagent was added to each well of the 96-well plate and incubated at 37 °C with 5% CO2 for 1–4 h. Cell viability was then assessed by measuring absorbance at 490 nm using a microplate reader SuPerMax 3100 (Flash, CN).
Statistics & Reproducibility
The data were obtained with randomization and blinding, with at least 3 biological replicates. Error bars represent means ± s.e.m. Differences between control and treatments were tested using two-tailed Student’s t-test. P values were obtained from two-tailed Student’s t-test: *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Data were analyzed using GraphPad Prism 9.5.1 software.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The deep sequencing data generated in this study have been deposited in the NCBI database under accession code PRJNA1238002. The raw data generated in this study are provided in the Supplementary Information/Source Data file. Source data are provided with this paper.
References
Chen, P. J. & Liu, D. R. Prime editing for precise and highly versatile genome manipulation. Nat. Rev. Genet. 24, 161–177 (2023).
Zhao, Z. et al. Prime editing: advances and therapeutic applications. Trends Biotechnol. 41, 1000–1012 (2023).
Gao, C. Genome engineering for crop improvement and future agriculture. Cell 184, 1621–1635 (2021).
Li, B. et al. Targeted genome-modification tools and their advanced applications in crop breeding. Nat. Rev. Genet. 25, 603–622 (2024).
Anzalone, A. V. et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157 (2019).
Nelson, J. W. et al. Engineered pegRNAs improve prime editing efficiency. Nat. Biotechnol. 40, 402–410 (2022).
Lin, Q. et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 38, 582–585 (2020).
Lin, Q. et al. High-efficiency prime editing with optimized, paired pegRNAs in plants. Nat. Biotechnol. 39, 923–927 (2021).
Chen, P. J. et al. Enhanced prime editing systems by manipulating cellular determinants of editing outcomes. Cell 184, 5635–5652.e29 (2021).
Anzalone, A. V. et al. Programmable deletion, replacement, integration and inversion of large DNA sequences with twin prime editing. Nat. Biotechnol. 40, 731–740 (2022).
Zong, Y. et al. An engineered prime editor with enhanced editing efficiency in plants. Nat. Biotechnol. 40, 1394–1402 (2022).
Liang, R. et al. Prime editing using CRISPR-Cas12a and circular RNAs in human cells. Nat. Biotechnol. 42, 1867–1875 (2024).
Martín-Alonso, S., Frutos-Beltrán, E. & Menéndez-Arias, L. Reverse transcriptase: from transcriptomics to genome editing. Trends Biotechnol. 39, 194–210 (2021).
Oscorbin, I. P. & Filipenko, M. L. M-MuLV reverse transcriptase: selected properties and improved mutants. Comput. Struct. Biotechnol. J. 19, 6315–6327 (2021).
Lee, J. et al. Prime editing with genuine Cas9 nickases minimizes unwanted indels. Nat. Commun. 14, 1786 (2023).
Shuto, Y. et al. Structural basis for pegRNA-guided reverse transcription by a prime editor. Nature 631, 224–231 (2024).
Jiang, F. et al. Structures of a CRISPR-Cas9 R-loop complex primed for DNA cleavage. Science 351, 867–871 (2016).
Khanna, K. K. & Jackson, S. P. DNA double-strand breaks: signaling, repair and the cancer connection. Nat. Genet. 27, 247–254 (2001).
Anand, R. et al. HELQ is a dual-function DSB repair enzyme modulated by RPA and RAD51. Nature 601, 268–273 (2022).
Li, X. et al. Development of a versatile nuclease prime editor with upgraded precision. Nat. Commun. 14, 305 (2023).
Peterka, M. et al. Harnessing DSB repair to promote efficient homology-dependent and -independent prime editing. Nat. Commun. 13, 1240 (2022).
Xu, X. et al. CircRNA inhibits DNA damage repair by interacting with host gene. Mol. Cancer 19, 128 (2020).
Conn, V. M. et al. A circRNA from SEPALLATA3 regulates splicing of its cognate mRNA through R-loop formation. Nat. Plants 3, 17053 (2017).
Su, X. et al. CircR-loop: a novel RNA: DNA interaction on genome instability. Cell Mol. Biol. Lett. 29, 89 (2024).
Liu, H. et al. Super enhancer-associated circRNA-circLrch3 regulates hypoxia-induced pulmonary arterial smooth muscle cells pyroptosis by formation of R-loop with host gene. Int. J. Biol. Macromol. 268, 13085 (2024).
Liu, C. X. & Chen, L. L. Circular RNAs: Characterization, cellular roles, and applications. Cell 185, 2016–2034 (2022).
Liu, B. et al. A split prime editor with untethered reverse transcriptase and circular RNA template. Nat. Biotechnol. 40, 1388–1393 (2022).
Feng, Y. et al. Enhancing prime editing efficiency and flexibility with tethered and split pegRNAs. Protein Cell 14, 304–308 (2023).
Litke, J. L. & Jaffrey, S. R. Highly efficient expression of circular RNA aptamers in cells using autocatalytic transcripts. Nat. Biotechnol. 37, 667–675 (2019).
Xi, D. C. et al. Helicase-assisted continuous editing for programmable mutagenesis of endogenous genomes. Science 386, eadn5876 (2024).
Arslan, S. et al. Engineering of a superhelicase through conformational control. Science 348, 344–347 (2015).
Gavrilov, M. et al. Engineered helicase replaces thermocycler in DNA amplification while retaining desired PCR characteristics. Nat. Commun. 13, 6312 (2022).
Yan, J. et al. Improving prime editing with an endogenous small RNA-binding protein. Nature 628, 639–647 (2024).
Kweon, J. et al. Engineered prime editors with PAM flexibility. Mol. Ther. 29, 2001–2007 (2021).
Bae, S., Park, J. & Kim, J. S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Durrant, M. G. et al. Bridge RNAs direct programmable recombination of target and donor DNA. Nature 630, 984–993 (2024).
Liu, Z. X. et al. Hydrolytic endonucleolytic ribozyme (HYER) is programmable for sequence-specific DNA cleavage. Science 383, eadh4859 (2024).
Eggers, A. R. et al. Rapid DNA unwinding accelerates genome editing by engineered CRISPR-Cas9. Cell 187, 3249–3261 (2024).
Landrum, M. J. et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–D868 (2016).
Acknowledgements
This work was supported by the Biological Breeding-National Science and Technology Major Project (2023ZD04074 to R.L.), the National Key R&D Program of China (2024YFA0917303 to R.L. and 2023YFF1001600 to K.C.), the Beijing Municipal Science & Technology Commission (Z241100009024035 to C.G.), and the National Natural Science Foundation of China (32388201 to C.G. and 32401242 to R.L.).
Ethics declarations
Competing interests
The authors declare the following competing interests: C.G. and R.L. have submitted a patent application on the prime editors developed in this work (Application No. 2024112204624). The remaining authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liang, R., Wang, S., Cai, Y. et al. Circular RNA-mediated inverse prime editing in human cells. Nat Commun 16, 5057 (2025). https://doi.org/10.1038/s41467-025-59120-7