
Mechanisms of Cadmium Neurotoxicity
镉神经毒性机制
SCI升级版 生物学2区SCI基础版 生物2区IF 4.9 如果4.9

作者:玛德琳·A·阿鲁巴雷纳 (
圣母大学神经科学与行为项目,圣母大学,IN 46556,美国
圣母大学化学与生物化学系,圣母大学,IN 46556,美国
信件应寄给的作者。
你。 J.莫尔。科学。 2023 , 24 (23), 16558; https://doi.org/10.3390/ijms242316558IF: 4.9 Q1 B2如果:4.9 Q1 B2
提交材料收到:2023年10月14日/修订:2023年11月17日/接受:2023年11月18日/发布:2023年11月21日
Abstract 抽象的
镉是一种重金属,对食品和饮料产品的污染日益严重。一旦摄入,镉就会产生毒性作用,对人类健康构成重大威胁。神经系统特别容易受到长期、低剂量镉的影响。这篇综述文章概述了镉的主要神经毒性机制。镉通过锌和钙转运蛋白进入神经系统,改变这些金属离子的稳态。一旦进入神经系统,镉就会通过减少 ATP 合成和增加活性氧的产生来扰乱线粒体呼吸。镉还通过增加神经递质释放的异步性和破坏神经递质信号蛋白来损害正常的神经传递。镉还会损害血脑屏障并改变糖原代谢的调节。总之,这些机制代表了多个位点的生化扰动,导致累积的神经系统损伤,从而增加神经系统和神经退行性疾病的风险。了解镉发挥其作用的方式对于制定针对镉引起的神经毒性损伤的有效治疗和预防策略至关重要。
1. Introduction 一、简介
镉是一种剧毒污染物,渗透到环境、工业和农业空间中。有毒物质和疾病登记局将镉列为对人类健康第七大危害物质[ 1 ],美国卫生与公众服务部于2021年将镉列为已知的人类致癌物[ 2 ]。最近的人类活动增加了人类对镉的接触。大多数商业镉是锌矿开采的副产品,用于电镀、电池生产、油漆颜料和塑料 [ 3 , 4 ]。这些活动将镉引入农业领域,植物很容易从受污染的土壤和水中吸收镉。此外,乙醇的镉污染很常见,在葡萄酒、啤酒、威士忌、杜松子酒和其他酒精产品中检测到的镉含量各不相同[ 5 ]。因此,普通人群最常见的接触源是受污染的食品和饮料产品[ 3 ]。
镉通过多种途径进入人体。摄入受污染的食品和饮料、吸入香烟烟雾中的雾化镉颗粒以及接触工业烟雾后嗅球中的颗粒积聚都会促进镉的吸收[ 6 , 7 ]。由于其非生物性质,镉没有内源性清除机制,因此尿排泄率较低。它在人体内蓄积,估计半衰期长达 23.5 年 [ 8 ]。由于这种积累,美国和欧洲未因职业接触镉的成年人体内的镉含量估计在 9.5 毫克至 40 毫克之间[ 9 ]。此外,人类的血液镉浓度约为 0.4 µg/L [ 10 ],脑脊液 (CSF) 镉浓度为 72 ng/L [ 10 , 11 ]。因此,人体脑脊液中的镉浓度仅比血液中的镉浓度低大约五倍。
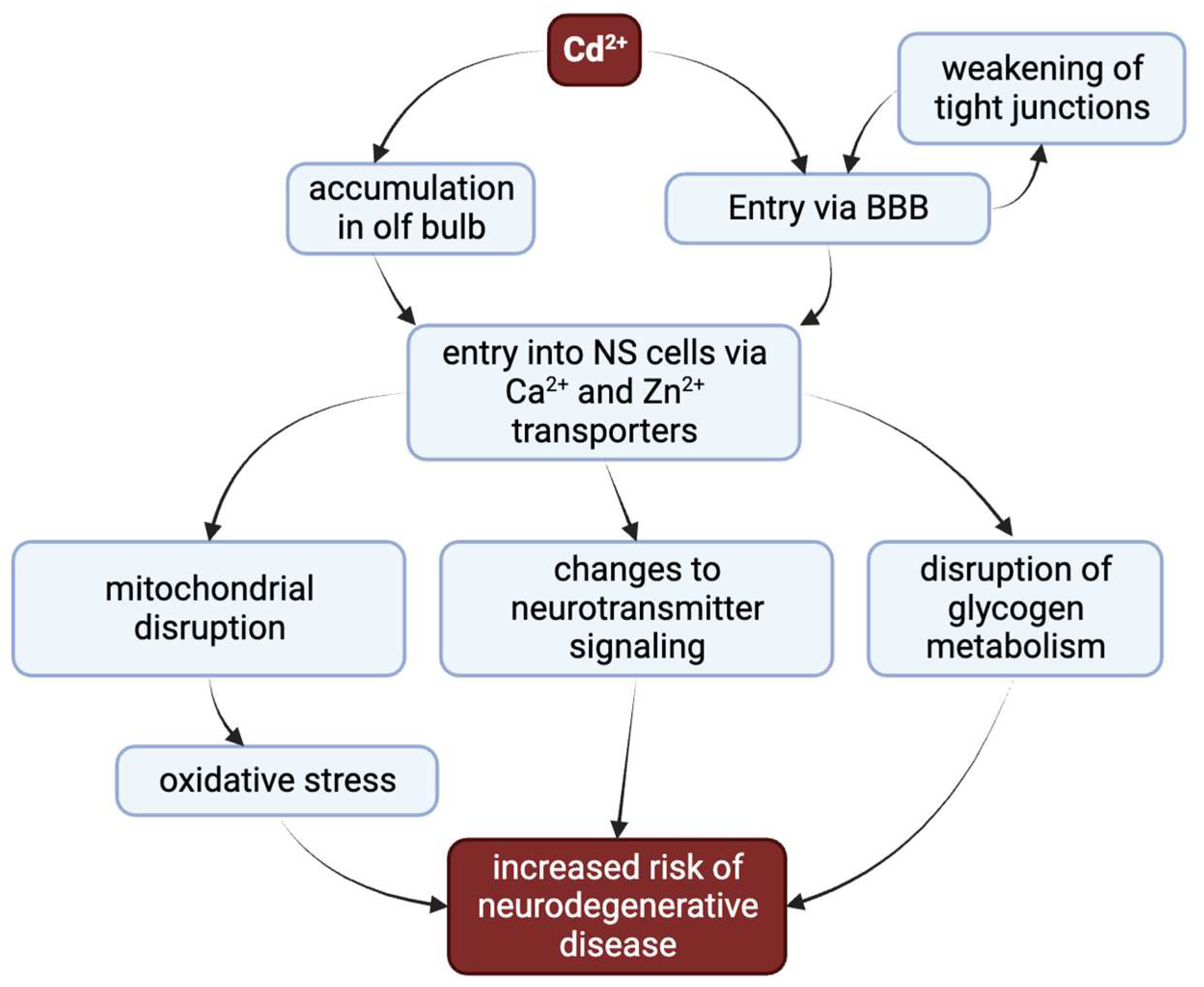
镉的慢性蓄积会导致多器官毒性,主要针对肾脏、骨骼、肝脏和神经系统[ 12 ],详见[ 9 ]。其中,神经系统是镉毒性特别脆弱的目标。镉会增加周围神经病变、平衡改变和视觉运动任务表现不佳的风险[ 13 ]。接触镉与老年人注意力下降、认知功能较差以及儿童学习成绩不良相关[ 13,14,15,16 ]。镉暴露还与阿尔茨海默病 (AD)、帕金森病 (PD) 和肌萎缩侧索硬化症( ALS ) 中观察到的神经退行性疾病病理相关[ 17,18,19 ]。镉通过多种方式发挥其神经毒性作用[12,20,21 ] (图1 ) 。在这里,我们回顾了有关导致神经系统功能障碍的外源性镉损伤部位的最新知识。

图 1.镉增加神经退行性疾病风险的多种主要途径。镉吸入后会积聚在嗅球中。当吸入或摄入时,镉会进入血液,通过削弱紧密连接来降低血脑屏障(BBB)的完整性。这使得镉进入神经系统组织。一旦进入神经组织,镉就可以通过选择其他二价阳离子的转运蛋白来有效地穿过细胞膜。神经毒性的主要机制是糖原代谢的破坏、神经递质信号传导的改变以及导致氧化应激的线粒体破坏。这些干扰共同增加了神经退行性疾病的风险。
2. Cadmium Entry to the Nervous System
2. 镉进入神经系统
镉主要通过口服进入神经系统,然后被吸收到血液中,并会损害血脑屏障(BBB)并在神经系统组织内积聚。镉吸入为神经系统提供了更直接的途径,因为嗅觉上皮缺乏血脑屏障提供的保护,允许镉直接吸收到神经组织中[ 22 ]。镉与参与神经元传递的生物必需金属阳离子类似,特别是钙和锌。镉、钙和锌主要是二价阳离子,具有相似的化学性质并有利于+2氧化态。钙和镉具有相似的离子半径(分别为 0.97 Å 和 0.99 Å)和电荷/半径比(Ca 2+ = 2.02 e/Å,Cd 2+ = 2.06 e/Å),从而赋予各自施加类似强静电的能力对生物大分子的作用力(综述于[ 23 ])。镉和锌是元素周期表第 IIB 族的元素,具有相同的电子构型,因此在离子-蛋白质相互作用中具有相似的化学行为。通过这种方式,镉可以利用内源性锌和钙特异性转运蛋白渗透神经系统细胞和细胞器。
多项研究表明镉是一种竞争性电压门控钙通道 (VGCC) 抑制剂 [ 24 , 25 , 26 ]。镉主要通过二氢吡啶敏感(L 型)VGCC 进入大鼠小脑颗粒神经元,因为它与 Ca 2+竞争通道孔内的空间。暴露于 100 µM 镉可阻止神经元去极化后胞质钙浓度的增加,并且镉能够渗透神经元。 N 型 VGCC 还与镉诱导的青蛙交感神经元 Ca 2+电流阻断有关 [ 27 ]。当Ca 2+通道主要打开时(0至+30 mV),镉在电压下完全且快速地阻断Ca 2+电流,表明N型VGCC是镉进入交感神经元的途径。由于 VGCC 密集地集中在突触前位点,因此突触前末梢是神经元细胞中镉摄取的一个显着位置([ 28 ]中有综述)。
镉还通过锌转运蛋白进入神经元细胞,其中最重要的是 ZIP6 和 ZnT3 转运蛋白 [ 28 , 29 ]。 ZIP6 是一种输入蛋白,位于海马锥体神经元的质膜上,而 ZnT3 是一种输出蛋白,大量存在于突触前神经元膜上,调节大脑的囊泡池 [ 30 , 31 ],详见 [ 22 ]。米穆纳等人。发现生命早期接触镉会增加大脑中镉的积累,增加 ZIP6 基因表达,并降低 ZnT3 表达[ 29 ]。 ZIP6 输入蛋白的同时上调和 ZnT3 输出蛋白的下调可能导致这些神经元中镉的积累。在后来的研究中,Mimouna 等人。研究了海马神经元中镉和 ZnT3 之间的相互作用。用氯化镉(0、0.5、5、10、25 或 50 µM)和氯化锌(0、10、30、50、70 或 90 µM)处理大鼠海马神经元 24 或 48 小时,下调 ZnT3 mRNA表达,锌的应用减弱了这种效应。在用 10 µM 和 25 µM 镉处理的细胞中,补充 30 µM 锌可显着改善镉诱导的神经毒性 [ 32 ]。据推测,镉和锌之间的物理化学相似性使得镉能够通过 ZnT3 进入突触小泡并积累,最终导致细胞死亡和神经元可塑性破坏。
3. Cadmium Effects on Mitochondrial Respiration
3. 镉对线粒体呼吸的影响
神经系统中的线粒体不仅在能量产生中发挥着关键作用[ 33 ],而且在神经元发育、功能和存活中也发挥着关键作用[ 34 ]。神经元是神经系统的功能单位,是 ATP 的高消耗者,因为它们不断需要维持动作电位传播所需的神经元浓度梯度、操作与囊泡循环相关的细胞机制、促进轴突运输并提供能量突触可塑性[33,34,35 ] 。因此,线粒体功能的任何破坏都可能导致能量不足,从而严重损害神经活动和健康。
氧化磷酸化依赖于强大的线粒体膜电位 (ΔΨm),以便通过 ATP 合酶产生 ATP [ 35 ]。嵌入线粒体内部基质中的电子传递链 (ETC) 利用电子携带分子的势能来产生强大的 ΔΨm。组成 ETC 的四种蛋白质复合物必须巧妙地处理氧化还原分子,以便适当地产生质子梯度,这是 ΔΨm 的基础。此外,作为 ETC 的副产品,会产生低浓度的活性氧 (ROS)。线粒体内的抗氧化剂分子(例如谷胱甘肽)可以缓解低水平的 ROS。然而,如果允许 ROS 增殖,无论是通过外部影响还是 ETC 的不当调节,所产生的氧化应激都会导致细胞损伤。
这种 ΔΨm 梯度可以通过线粒体解偶联蛋白进行调节,线粒体解偶联蛋白可以响应细胞的能量需求、保持一致的温度或控制渗透膨胀。然而,各种病理状况会破坏 ΔΨm,导致线粒体呼吸受损。例如,氧化应激等因素可以触发线粒体通透性转换孔(PTP)开放,从而导致 ΔΨm 去极化[ 36 ]。这种去极化会抑制 ATP 合成并损害线粒体的整体功能。
线粒体的重要性在神经退行性疾病(包括 AD、PD 和 ALS)的背景下变得最为明显。这些病症的特点是线粒体功能障碍[ 35 ]。异常包括能量产生受损、ROS 产生增加和钙处理受损,共同导致神经元变性和这些疾病的临床表现[ 36 ]。
3.1. Cadmium Interference with the Electron Transport Chain
3.1.镉对电子传输链的干扰
线粒体已成为镉毒性的主要目标(有关专门关注该主题的优秀评论,请参阅[ 37 ])。啮齿动物模型中的实验证据支持了这一点,其中小鼠肝脏分离线粒体的镉暴露导致广泛的细胞器损伤[ 38 ]。镉破坏线粒体功能的一种机制是干扰 ETC 内的特定蛋白质复合物,从而减少 ΔΨm,从而削弱驱动 ATP 合成的质子动力。镉与 ETC 复合物 I 在 Q 结合位点和 NADH 结合位点相互作用,降低复合物 I 穿梭电子和传输质子以产生和维持 ΔΨm 的能力。镉与复合物 III 的 Q o位点的相互作用将 ROS 的产生重定向到膜间空间,有效地绕过基质抗氧化防御 [ 39 , 40 ]。通过破坏复合物 I 和 III 的正常功能,由此产生的 ΔΨm 减少最终导致有效合成 ATP 的能力下降,并增加破坏性胞质 ROS。
3.2. Cadmium Opens the Permeability Transition Pore
3.2.镉打开渗透性转变孔
此外,镉会诱导渗透性过渡孔(PTP)的打开,这是一种位于线粒体内外隔室之间界面的动态蛋白质复合物[ 41 ]。 PTP 允许小分子通过线粒体内膜扩散,消散 ΔΨm,从而停止 ATP 合成。 PTP 的打开还通过释放细胞色素 C 储备来充当细胞凋亡的信号。线粒体内部梯度本身的减弱可以在前馈机制中触发 PTP 的打开,从而导致最终的细胞死亡。目前尚不清楚镉在上述机制中通过削弱 ΔΨm 在多大程度上打开 PTP,或者镉是否直接与 PTP 本身相互作用以增加打开的可能性,或两者兼而有之。
有一些证据表明镉直接与 PTP 相互作用以增加开口,与镉对 ΔΨm 的影响无关。镉与 PTP 复合物的成分、腺嘌呤核苷酸易位子 (ANT) 在半胱氨酸残基上的硫醇基团处相互作用,可能导致 ANT 功能的改变 [ 39 ]。 ANT 交换胞质 ADP 和基质 ATP,使胞质 ATP 从线粒体输出,同时将 ADP 输送到线粒体 [ 39 , 40 ]。结构研究表明,ANT 蛋白的 ADP/ATP 交换是通过“诱导过渡拟合”模型发生的。该过程始于 ADP 在“c 状态”的结合,此时蛋白质完全向膜间空间开放。这种结合触发构象转变为“m 状态”,此时蛋白质完全向线粒体基质开放,促进 ADP 与 ATP 的交换 [ 42 ]。抑制 ANT 可阻断镉诱导的 PTP 开放 [ 43 ]。这种镉诱导的 PTP 打开也可以通过添加线粒体钙输入抑制剂来阻断,这表明镉正在通过这种转运蛋白进入线粒体内部 [ 39 ]。
此外,钙可以通过环孢素 A (CsA) 依赖性机制诱导 PTP 打开 [ 43 ],而镉则独立于钙-CsA 途径打开 PTP。由于钙的增加是神经元内细胞凋亡的有效细胞内信号,因此镉绕过钙诱导的细胞凋亡机制的能力代表了不受调节的神经元死亡的另一种途径。这种不依赖于 CsA 的凋亡途径的调节机制尚不清楚。
3.3. Recent Focuses of Mitochondrial Apoptosis/Dysfunction: ER Stress and SIRT1
3.3.线粒体凋亡/功能障碍的最新焦点:ER 应激和 SIRT1
越来越清楚的是,线粒体应激和内质网(ER)应激之间的关系都会导致镉的毒性作用。根据多项研究,镉暴露可能会引起内质网和线粒体应激反应之间的串扰,最终导致细胞凋亡 [ 44 , 45 ]。这种串扰的一个重要方面是促凋亡蛋白 Bim 和 Bax 的参与,它们在急性镉暴露后表达上调。特别是 Bax 从细胞质转移到线粒体,导致线粒体凋亡。此外,镉暴露会导致细胞色素c从线粒体释放到细胞质。这种释放触发细胞凋亡信号并激活半胱天冬酶,导致细胞凋亡[ 44,46,47 ]。
此外,人类细胞系暴露于镉会导致细胞内ROS水平以剂量依赖性方式增加。这一代 ROS 以前馈方式发生,最终诱导 GADD153,这是一种启动细胞死亡的标记物 [ 45 ]。针对这一过程的保护措施是抗氧化剂白藜芦醇,它抑制 ER 应激和 GADD153 并激活 Sirtuin1 (SIRT1) [ 48 ]。
最近的研究还指出 SIRT1 是氧化应激生化反应的关键调节因子 [ 45 ]。 SIRT1 是一种烟酰胺二核苷酸 (NAD + ) 依赖性脱酰酶,以其调节 DNA 修复、炎症、脂肪酸氧化、脂肪分化等细胞过程的能力而闻名 [ 48 ]。镉对 SIRT1 的抑制导致神经元细胞内氧化应激显着增加。随之而来的氧化应激会破坏线粒体功能,最终导致神经细胞死亡。这种现象已在 PC12 细胞(一种神经元样细胞系)和原代大鼠大脑皮层神经元中观察到[ 45 ]。激活 SIRT1 可以防止 ROS 的积累和细胞损失,并阐述了 SIRT 激活剂影响 SIRT1 活性的潜在机制,特别是通过去乙酰化 PGC-1a。这种脱乙酰化被认为有助于增强氧化代谢,在细胞对氧化应激的反应中发挥着至关重要的作用[ 49 ]。 SIRT1 在结构上对神经系统很重要,因为它促进轴突伸长、神经突生长和树突分支。此外,它被发现对于记忆形成及其针对阿尔茨海默氏症、帕金森氏症和运动神经元疾病等神经退行性疾病的保护措施至关重要[ 50,51,52,53 ]。
线粒体相关内质网膜 (MAM) 中镉诱导的神经毒性的确切分子机制仍不清楚。 MAM 由多种蛋白质组成,包括线粒体融合蛋白 2 (Mfn2)、电压依赖性阴离子通道 (VDAC) 和葡萄糖调节蛋白 75 (Grp75)。这些蛋白质通过 ER 上的肌醇 1,4,5-三磷酸受体 (IP3R) 和线粒体上的电压依赖性阴离子选择性通道蛋白 (VDAC) 促进钙离子从 ER 转运至线粒体 [ 49 ] 。暴露于镉会增加 Mfn2、Grp75 和 VDAC1 的表达[ 52 ]。此外,当镉处理导致 Mfn2 被敲除时,PC12 细胞和原代神经元的线粒体钙吸收均显着减少。值得注意的是,这揭示了镉诱导神经元细胞自噬的主要驱动因素可能是 MAM 促进的线粒体钙的摄取,特别是 IP3R-Grp75-VDAC1 复合物受 Mfn2 调节。 Mfn2 与 IP3R-Grp75-VDAC1 复合物的运作之间的相互作用代表了对镉暴露后线粒体功能障碍的理解的突破 [ 50 ]。
3.4. Cadmium-Induced Autophagy
3.4.镉诱导的自噬
自噬是一种受调节的细胞死亡形式,涉及一系列将目标材料引导至溶酶体进行回收的步骤(有关神经退行性疾病中自噬的广泛综述,请参阅[ 51 ])。镉诱导的自噬与神经退行性疾病有关,尽管这种关系的性质仍然存在一些争议[ 52,53,54 ]。因此,本节重点介绍最近发表的有关镉诱导自噬主题的论文。
由于自噬是消除过量蛋白质的关键过程,因此人们认为,通过镉破坏自噬过程可能会导致过量的错误折叠蛋白质,从而导致神经退行性疾病[ 12 ]。镉通过增加自噬体形成来触发神经元凋亡,其标志是神经元细胞中 LC3-II 和 p62 升高,从而导致神经元凋亡 [ 55 ]。药物雷帕霉素可防止镉诱导的 LC3-II 和 p62 增加。镉诱导的细胞凋亡依赖于自噬体的过量产生,通过阻止自噬体-溶酶体融合[ 55,56,57 ]。然而,最近的其他研究表明,镉在细胞培养模型中通过钙依赖性激活 JNK 信号通路来抑制自噬 [ 58 ]。
最近的研究推进了预防镉诱导的自噬通量变化的改善策略的研究。鹅委陵菜是一种原产于中国青藏高原的草本植物,以其营养丰富和在中药中的应用而闻名。新兴研究强调委陵菜多糖(PAP)是这种草药的主要生物活性成分,可作为预防氧化应激、线粒体细胞死亡和细胞凋亡的候选药物[59,60,61,62 ] 。 PAP 通过抑制 PI3K III 类/Beclin-1 信号通路,可能通过自噬减轻镉诱导的神经元死亡 [ 63 ]。有趣的是,增加自噬的药物似乎也有望预防镉引起的神经毒性损伤。利格列汀是 FDA 批准的用于治疗 2 型糖尿病的抗糖尿病药物,也显示出针对认知能力下降的神经保护作用 [ 64 , 65 ]。利格列汀对大鼠镉暴露的神经保护作用的研究表明,利格列汀可以预防镉引起的认知缺陷。然而,利格列汀刺激海马 AMPK/mTOR 通路,从而对自噬进展产生积极影响。据认为,自噬的增加刺激了神经元错误折叠蛋白的清除,从而改善了这种情况下的认知障碍[ 66 ]。总之,这些结果表明需要进一步探索镉在自噬过程中的作用。
4. The Role of Cadmium in Synaptic Transmission
4. 镉在突触传递中的作用
突触本身是镉毒性的脆弱目标。为了有效传输神经元信号,生物金属阳离子必须与一系列电压门控和配体门控通道协同作用。镉与这些离子(特别是钙和锌)的物理化学相似性,使其在突触水平具有神经毒性,因为镉渗透到突触前神经元,诱导氧化应激,并最终加剧神经元变性。
4.1. Cadmium-Induced Asynchronous Neurotransmitter Release
4.1.镉诱导的异步神经递质释放
神经递质释放的同步是有效神经通讯的标志。神经递质的释放发生在动作电位后数百毫秒内,以确保神经元之间的精确通信[ 28 ]。事实上,一些研究已将异步释放与 AD、脊髓性肌萎缩症 (SMA) 和 ALS 中的神经退行性疾病病理联系起来 [67,68,69,70 ] 。镉可能会增加异步神经递质释放,进一步加剧这些神经退行性疾病的病理。
在青蛙神经末梢的远端室中应用 0.1 µM 去同步神经递质释放的镉。这种异步性伴随着线粒体活性氧产生和脂质过氧化的急剧增加,表明镉诱导的氧化应激与这种去同步性同时发生。给予抗氧化剂和 NADPH 氧化酶抑制剂可以完全阻断去同步化[ 55 ]。这种异步性的一种可能机制依赖于镉除了作为氧化应激引发剂的作用之外还作为 VGCC 拮抗剂的作用。细胞外镉可能取代天然钙作为流经 L 型 VGCC 的金属离子。镉存在下钙内向电流的减少导致胞质钙的突触前尖峰减弱,这对于囊泡机械的协调是不可或缺的。因此,在存在镉的情况下,VGCC 必须保持较长时间的开放状态,以允许足够的钙流入,从而启动钙依赖性突触前过程。 VGCC 长时间开放可能会延长去极化动作电位到达和神经递质释放之间观察到的延迟,从而解释了观察到的异步性。
4.2. Cadmium Disruption of Neurotransmission
4.2.镉对神经传递的干扰
除了延迟神经递质释放外,镉还会破坏突触小泡内的神经递质包装,减少每次释放事件可用的神经递质数量。在谷氨酸能神经元中有特别的证据表明这一点。囊泡转运蛋白依靠 V-ATP 酶产生的质子电化学梯度将神经递质包装到囊泡中 [ 71 , 72 ]。分离的 Wistar 大鼠突触体中 50 µM 的镉会导致将谷氨酸包装到突触囊泡中所需的质子梯度消散,导致去极化引起的谷氨酸胞吐作用减少,并降低细胞外谷氨酸浓度 [ 73 ]。尽管没有直接观察到 V-ATP 酶被破坏的机制,但与 V-ATP 酶半胱氨酸残基中的硫醇基团的相互作用可能是罪魁祸首。
此外,已观察到镉暴露会引起胆碱能毒蕈碱受体和乙酰胆碱酯酶(AChE)变体的变化[ 74 ]。具体而言,Cd 2+暴露记录了 AChE-S(突触变体)的基因表达升高,同时降低了 AChE-R(通读变体)的基因表达。 AChE 变体的这种修饰与这些神经元的细胞死亡有关。此外,镉治疗会破坏毒蕈碱受体,特别是 M1 和 M3 受体,它们在调节记忆和学习过程中发挥着至关重要的作用。这种对受体的干扰可能会导致接触镉后观察到的认知障碍。尽管镉改变毒蕈碱受体和乙酰胆碱酯酶变体的精确机制尚未完全阐明,但氧化应激已被认为是这一过程中的潜在中间因素。
5. Cadmium and Other Metals
5. 镉和其他金属
5.1. Cadmium Disruption of Zinc Signaling and Homeostasis
5.1.镉破坏锌信号传导和体内平衡
锌本身可以防止镉引起的海马神经毒性[ 25 ],在浓度为 25 µM 时,会减少量子释放并显着使神经递质释放不同步。锌可以在细胞系统中充当促氧化剂或抗氧化剂,过量和缺乏都会导致氧化应激([ 75 ]中综述)。锌诱导的氧化应激与培养的皮质神经元 [ 44 , 47 ] 和 AD [ 45 ] 中的神经变性和细胞死亡有关。镉通过锌转运蛋白的流入可能会破坏这种锌的动态平衡,导致氧化应激加剧。因此,锌和镉可能协同作用,诱导突触前末梢氧化应激,最终导致量子释放减少和异步,从而促进神经退行性变。
镉暴露导致锌转运蛋白 ZnT3 下调,导致下游效应,影响大脑中的关键信号通路。这种下调会引发级联反应,减少海马脑源性神经营养因子原肌球蛋白受体激酶 B (BDNF-TrkB) 和 Erk1/2 信号传导,这些细胞内信使在神经元可塑性和生长中发挥着不可或缺的作用 [ 50 ]。 TrkB 神经营养蛋白受体和随后的 BDNF 激活对于促进神经元可塑性至关重要,抗抑郁药与神经营养蛋白受体(特别是 TrkB)的结合先前已被证明可促进 BDNF 激活并启动神经元可塑性 [ 51 ]。虽然与 TrkB 神经营养蛋白受体结合的抗抑郁药通过激活 BDNF 来帮助神经元可塑性,但镉等其他外源物质可能通过 ZnT3 下调等间接机制来抑制它。需要进一步的研究来阐明镉阻碍神经元可塑性的机制。
神经元衰老是累积细胞损伤的标志。然而,神经元衰老的机制多种多样且复杂。多项研究表明氧化应激和神经元衰老密切相关[ 48,58,59 ]。过量活性氧的存在会导致蛋白水解,从而影响细胞功能并表现为衰老。加芬克尔提出了“衰老的锌假说”[ 76 ]。根据这一假设,饮食中缺锌会导致其金属酶的锌利用率减少,从而导致金属酶失调。这种失调因细胞类型而异,会导致蛋白质畸形和积累,最终表现为衰老。他后来预测锌缺乏是继发于镉毒性的[ 77 ]。根据这一假设,在对照模型和神经退行性模型中,镉可能会导致锌功能障碍并最终催化神经元衰老。
2020 年,谢等人。回顾了锌在阿尔茨海默病发展中的作用,解释说锌稳态的破坏可能对阿尔茨海默病有影响。在中枢神经系统中,ZnT3 将锌包装到集中在海马体、杏仁核和大脑皮层的锌能神经元的突触前囊泡中。锌能神经元突触前释放锌被认为可以调节神经元可塑性以及学习和记忆[ 78 ]。然而,AD 患者死后脑组织分析显示 ZnT3 的 mRNA 和蛋白质水平降低 [ 79 , 80 ]。 ZnT3 下调会阻止锌包装到囊泡中,导致神经元内细胞内锌过量,很容易与与 AD 发病机制相关的淀粉样蛋白-β (Aβ) 寡聚物结合。锌与 Aβ 的结合改变了 Aβ 的二级结构,从而促进神经毒性球形物质的形成 [ 60 ],同时也限制了锌作为可塑性调节剂的生物利用度。 AD 患者血清中表现出锌缺乏,这可能是由于锌和 Aβ 之间的持续相互作用促进了神经毒性低聚物和原纤维的形成,从而有效地阻碍了锌发挥其天然生物学作用。
5.2. Cadmium Contributions to Metal Imbalance in Alzheimer’s Disease
5.2.镉导致阿尔茨海默病金属失衡
除镉外,还有几种痕量金属与 AD 发病机制和进展有关([ 81 ] 中综述)。镉引起的锌失调可能是加重和进展阿尔茨海默病的危险因素。尽管已有研究探讨锌与阿尔茨海默病之间的联系[82,83,84 ] ,但人们对镉、锌与神经元衰老之间的关系知之甚少。在AD患者中观察到生物必需金属离子,特别是锌、铜和铁的不平衡[ 85 ]。在 2023 年对 73 项测量 AD 患者微量元素水平的研究进行的荟萃分析中,Li 等人。报道了血清中铜、血浆中铁和头发中锌水平的变化[ 86 ]。尽管李等人。没有解决镉含量问题,2017 年对 AD 患者循环中有毒金属的荟萃分析报告称,与对照组相比,镉含量有所增加 [ 87 ]。上述每种金属离子失衡都会对神经变性产生影响,它们复杂、独特的相互作用可能表现为 AD 病理。
铜、铁和锌的失衡加上外源性镉暴露似乎会诱发氧化应激加剧的循环,并促进有毒 Aβ 的形成。如前所述,镉通过锌转运流入会破坏锌稳态,加剧氧化应激,并下调 ZnT3 转运蛋白 [ 88 ]。在 AD 患者中也观察到 ZnT3 的下调,表明锌失衡与外源性镉暴露密切相关 [ 64 , 65 ]。未被 ZnT3 隔离到囊泡中的过量细胞内锌可能很容易与 Aβ 单体结合,促进有毒低聚物的产生 [ 60 ]。最近探索铜水平与 AD 之间关系的研究得出了不同的结果。一些分析报告称,与对照组相比,AD 患者的铜水平有所增加,而其他研究报告称,铜水平没有显着变化 [ 86 ]。然而,铜,特别是 Cu(II),已被观察到与 Aβ 结合并导致大脑中的斑块,最终加剧氧化应激和神经炎症,就像外源性镉和锌失衡会导致氧化应激一样[ 89 ]。铁还与 Aβ 单体结合,使这些单体发生结构变化,从而促进有毒 Aβ 寡聚物的形成。铁通过 Aβ 单体 N 端区域的 3 个组氨酸残基和 1 个酪氨酸残基与 Aβ 结合,从而减少 Aβ 的螺旋结构并增加 β 片层含量 [ 90 ]。 这种结构改变会促进有毒 Aβ 寡聚体的形成,并加剧神经炎症,从而可能加剧铁失衡和氧化应激,正如在镉、锌和铜中观察到的那样 [ 91 ]。镉、锌、铜和铁都与 AD 的进展有关。每种物质的不平衡都会导致氧化应激和神经炎症的永久循环,这有待进一步研究,并最终导致有毒 Aβ 寡聚物的产生,这是 AD 病理学的一个标志。
6. Cadmium and the Blood–Brain Barrier
6. 镉与血脑屏障
BBB 拥有高度特异性、严格调节的极性上皮细胞结构,主要依靠紧密连接 (TJ) 的形成来控制脑血管系统和神经系统细胞外液之间的通透性。跨膜和膜相关细胞质蛋白的集合包含 TJ ,并起到控制被动扩散的作用,限制极性溶质进入 CNS 并在解剖学上将 CNS 组织与血流分离[ 20,92,93 ]。 TJ 还受到周细胞、血管周围小胶质细胞、星形胶质细胞和神经元的调节。 BBB 的集体结构和调节元件被称为“神经血管单元”(NVU),该术语于 2001 年引入[ 94 ]。然而,研究报告称,镉对血脑屏障造成了破坏性改变,这可能是 AD、PD 和慢性创伤性脑病等神经退行性疾病的病理生理学基础,尽管确切的分子机制尚不清楚 [ 20 , 95 ]。
尽管镉很容易穿过幼年动物的未成熟血脑屏障,但它通常会受到严格的 TJ 调节而无法穿过成年动物的血脑屏障 [ 96 ]。然而,成人大脑中确实会发生积累,特别是当镉与允许穿过血脑屏障的载体(例如乙醇)结合时[ 97 , 98 ],乙醇通常含有痕量的镉作为掺杂物[ 5 ]。有趣的是,乙醇启动生化级联反应,以与镉类似的方式改变 BBB 的渗透性,两者都是从间接上调 ROS 开始,导致细胞应激反应,最终导致 NVU 蛋白表达减少 [ 97 , 99 , 100 ]。
布兰卡等人。报道称,用 10 µM 氯化镉 (CdCl 2 ) 处理大鼠脑内皮细胞系 (RBE4) 后,氧化应激的诱导迅速发生,ROS 的产生介导内质网 (ER) 信号通路,最终导致紧密连接的结构破坏和细胞骨架 BBB 蛋白 [ 100 ]。暴露后,ROS 过量产生在 5 分钟时达到峰值,然后在 10 分钟时恢复到正常水平,并在两小时后再次增加,表明急性镉施用后可能存在双重短期和长期氧化应激反应。氧化应激也会激活内质网应激反应,作者对 GRP78(一种经过充分研究的表明内质网应激的分子伴侣蛋白)的研究证明了这一点,并发现与对照组相比,CdCl 2暴露使 GRP78 表达增加了三倍。应激反应后,暴露后 8 小时测量到的凋亡蛋白 caspase-3 显着上调,并且构成 BBB 紧密连接和细胞骨架结构的三种蛋白的免疫细胞化学染色异常:闭锁小带-1 (ZO-1) ) 蛋白质、丝状肌动蛋白微丝 (F-肌动蛋白) 和波形蛋白。 ZO-1 表现出免疫细胞化学染色缺失,并且 F-肌动蛋白可见应力纤维形成。还观察到波形蛋白的破裂和拉伸。通过氧化应激依赖性内质网应激反应破坏这些蛋白质的特征是 TJ 破坏,最终导致中枢神经系统继发性损伤,类似于神经退行性疾病中观察到的情况。
2021 年,Zhang 等人。假设镉破坏 BBB 结构的扩展机制 [ 99 ]。将转基因斑马鱼胚胎暴露于 CdCl 2 (0、10、50、100 或 500 µM)中,通过破坏内皮细胞-细胞粘附并以剂量依赖性方式诱导脑出血,改变了 BBB 形态。 BBB 蛋白 ZO-1、血管内皮钙粘蛋白 (VE-cadherin) 和 F-肌动蛋白的定位和功能改变是由于镉诱导的氧化应激级联反应所致。然而,这种氧化应激介导了蛋白酪氨酸磷酸酶 (PTPase) 的抑制,PTPase 是一种调节 BBB 完整性的酶 [ 101 ]。据观察,氧化应激可介导人鼻上皮细胞中 PTPase 抑制导致蛋白质酪氨酸过度磷酸化引起的紧密连接损伤[ 102 ]。在斑马鱼模型中,PTPase 的抑制还导致 VE-钙粘蛋白和 ZO-1 的磷酸化迅速增加,从而开始将它们从典型的 BBB 结构中取代。 PTPase 的抑制导致 BBB 严重破坏和 occludin 蛋白水解,解释了治疗后 48 小时胚胎表现出的 BBB 通透性增加以及随后的脑出血。镉诱导的 BBB 结构破坏是氧化剂诱导级联反应的结果,多种下游分子机制与 TJ 和细胞骨架破坏有关。需要进一步的研究来阐明这些机制之间可能的相互作用,这些机制有助于在外源性镉暴露后观察到的全面 BBB 破坏。
如果我们不承认血脑屏障扰动和氧化应激本身都会加剧神经炎症,那就太失职了,神经炎症越来越被视为多种神经系统疾病的致病因素,特别是与神经退行性疾病有关。尽管篇幅限制了对神经炎症及其在整个生命周期中的有害影响的充分讨论,但该主题已在其他地方得到了很好的评论[ 103、104、105、106、107 ]。然而,镉相关神经炎症值得讨论的一个关键方面是镉对小胶质细胞的激活。小胶质细胞在大脑中表现出类似巨噬细胞的功能,包括向 T 细胞呈递抗原、一般免疫监视以及促炎细胞因子(如 TNF-α、IFN-γ 和 IL-6)的分泌[ 108 ]。镉可以通过产生 ROS 和增加 NF-κB(一种参与炎症反应的转录因子)的表达以及上调 caspase-3(一种参与神经元细胞凋亡的蛋白质)来激活小胶质细胞过度损伤的促炎症功能 [ 109 、 110 、 111 ]。
7. Cadmium’s Effects on Glycogen Metabolism
7. 镉对糖原代谢的影响
大脑中的糖原代谢对于重要的中枢神经系统功能至关重要。大脑的能量消耗非常高,一项研究声称它占成人全身静息葡萄糖消耗的 20-25%。大脑的发育可能需要更大比例的这种能量[ 112 ]。虽然糖原是满足神经元高能量需求的容易获得的葡萄糖来源所必需的,但糖原过多会导致神经变性,最常见于糖原贮积病[ 113,114,115 ]。因此,糖原代谢和储存的损害对生物体的神经功能特别有害。由于镉干扰细胞糖原途径,糖原失调是其神经毒性的一个主要途径。
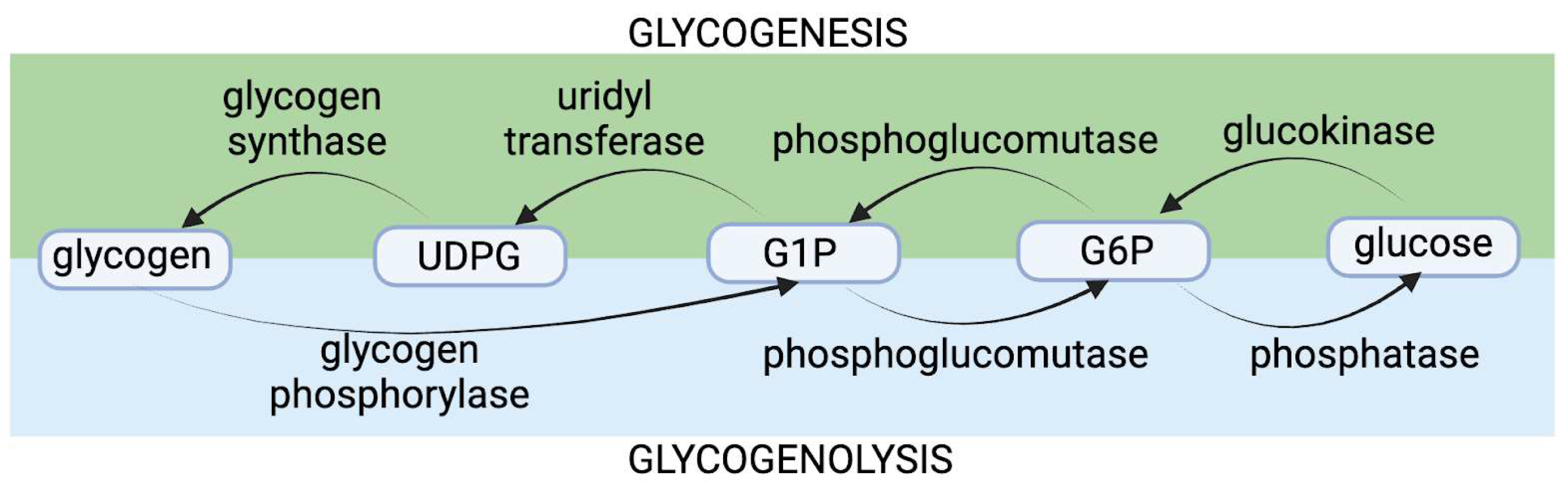
从历史上看,镉对糖原具有神经毒性的假设是基于镉对糖原磷酸化酶(GP)的功能性损害,该酶通过促进糖原多糖中葡萄糖-1-磷酸(G1P)单糖的裂解来催化糖原分解的第一步糖原分解作用[ 116 ](图2 )。糖原磷酸化酶在人体中以三种同工酶的形式存在,其中脑糖原磷酸化酶(bGP)是中枢神经系统主要的促进糖原分解的GP同工酶。神经元表达这种 bGP 酶,星形胶质细胞表达 bGP 和肌肉 GP 同工酶 [ 117 ]。神经元含有可测量量的糖原,但邻近的神经胶质星形胶质细胞是中枢神经系统的主要含糖原细胞[ 115 ]。神经元中的糖原积累实际上可能是神经退行性变的标志[ 118 ]。早期研究表明,镉的神经毒性是由于糖原过度积累所致。虽然有一些优点和证据支持这一最初的假设,但这一想法可能不准确,或者仅代表了镉糖原相关神经毒性的一部分。最近的证据表明,镉糖原相关神经毒性的主要机制是糖原储备的消耗,而不是糖原的积累。

图 2.糖原分解是细胞储存的糖原分解为葡萄糖的代谢过程。糖生成是相反的过程,葡萄糖聚合成糖原。如图所示,糖原分解和糖原生成都是由关键酶介导的。
7.1. Cadmium and Glycogen Phosphorylase Impairment: The Original Hypothesis
7.1.镉和糖原磷酸化酶损伤:最初的假设
据推测,镉会损害 bGP 的功能,导致神经系统细胞中糖原的积累 [ 116 ]。这一假设的基础源自研究表明 bGP 中半胱氨酸残基的硫醇基团对镉等金属离子敏感 [ 116 , 119 ]。由于糖原分解抑制,星形胶质细胞中糖原的积累可能是镉暴露引起神经系统症状的重要机制。
其他糖原磷酸化酶抑制剂(例如 CP-91149、CP-320626 和黄吡醇)已在杀死癌细胞的背景下进行了研究[ 120 ]。由于这种酶促损伤,糖原分解预计会被阻断,这意味着细胞无法将葡萄糖循环到磷酸戊糖途径中,并最终被细胞凋亡消除。因此,镉诱导的糖原介导的神经毒性的最初假设是,镉干扰半胱氨酸残基上的bGP结构,降低酶功能,并阻断糖原分解,导致糖原储存不当,最终导致神经细胞死亡。
这一假设得到了一些实验证据的支持。例如,在大鼠中,发现浓度为 0.3 毫克/千克体重的醋酸镉 (CdAc 2 ) 中毒会破坏糖酵解酶的功能,导致皮下注射 CdAc 2时糖原积累增加 20%每周两次,持续三个月[ 121 ]。引人注目的是,本研究中使用的 0.3 mg/kg 浓度完全在典型成人体内镉含量的估计范围内(0.12 mg/kg 至 0.5 mg/kg)[ 9 ]。此外,已发现,在每日注射直至孕龄之前,CdCl 2浓度为0.49 mg/kg怀孕Wistar大鼠体重的镉暴露会增加大鼠胎盘中糖原的积累[ 122 ]。
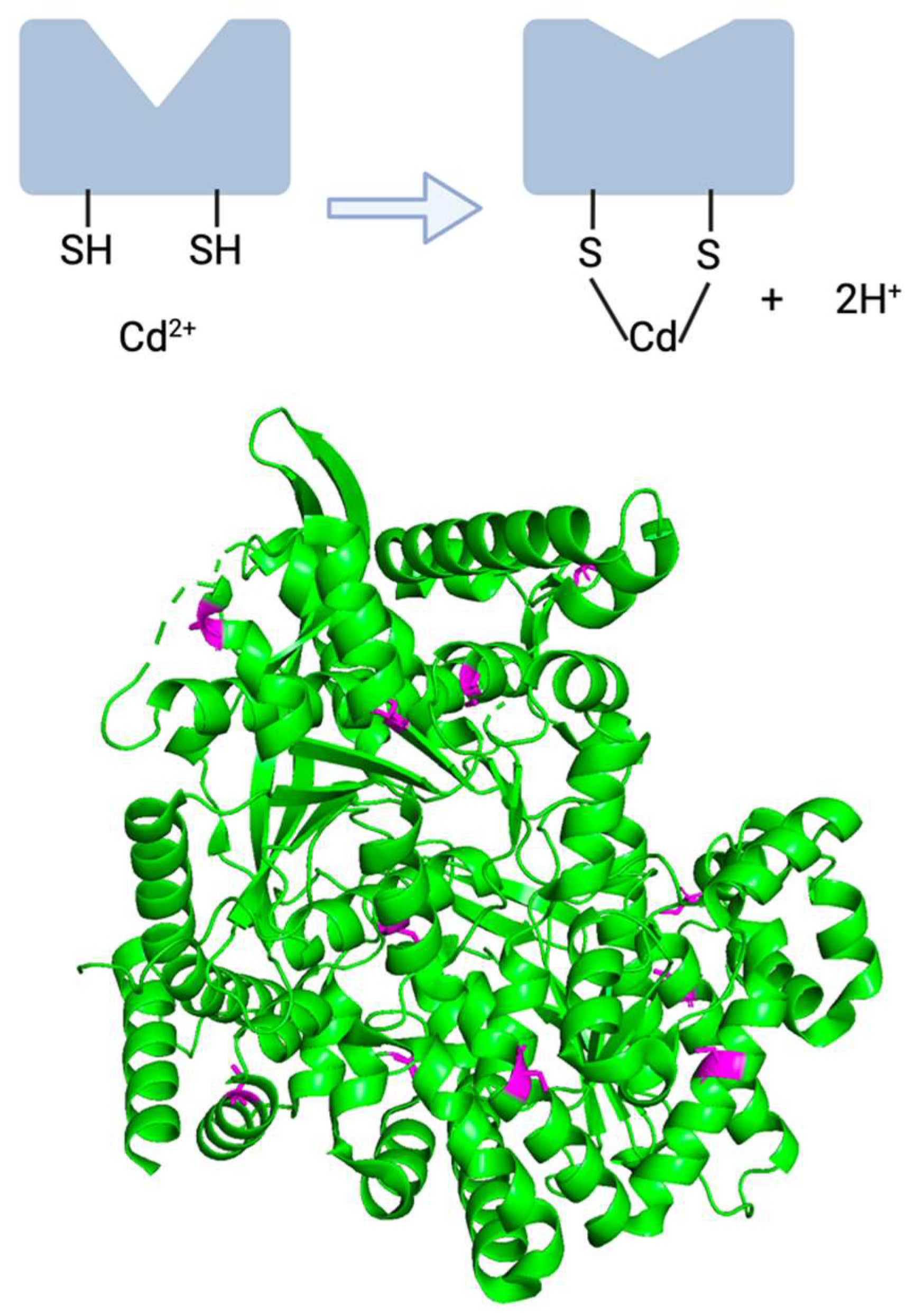
有关 bGP 酶本身的信息也证实了镉可能干扰关键半胱氨酸残基,最终破坏酶功能的观点。由于重金属对巯基/硫醇基团的高亲和力,镉等重金属能够破坏依赖半胱氨酸的酶的功能 [ 17 , 90 ](图 3 (上))。人类 bGP 的一级结构揭示了该酶有 14 个重要的半胱氨酸残基,这一点值得注意,因为 8% 的人类蛋白质没有一个半胱氨酸残基 [ 92 , 123 ]。此外,bGP的晶体结构已于2016年以高分辨率(2.5 Å和3.4 Å)得到解析,这可能有助于进一步阐明这一想法[ 113 ]。该结构突出了 bGP 半胱氨酸残基的扩散性质以及天然结构多个基序中硫醇基团的相关性(图 3 (底部))。因此,由于 bGP 的半胱氨酸具有多个硫醇基团以及镉进入神经系统的能力,bGP 的损伤可能是镉干扰其易受影响的半胱氨酸残基的结果。值得注意的是,这些半胱氨酸残基既不位于调节剂 AMP 的催化位点,也不位于调节剂 AMP 的变构结合位点,尽管镉诱导的蛋白质其他位置半胱氨酸残基的修饰可能会对酶活性产生负面影响。这种干扰发生的可能机制如图 3所示,其中对 bGP 三维结构重要的未氧化半胱氨酸改为与镉阳离子配位。

图 3. (上)镉可以与半胱氨酸残基的巯基相互作用,导致酶结构发生变化。 (底部)bGP 蛋白质结构,突出显示半胱氨酸残基(PDB 条目:5IKP)。
人类临床数据也支持有关糖原积累介导的神经毒性的理论。例如,在 IX 型糖原贮积病的情况下,糖原积累是由糖原磷酸化酶失活引起的,并已被观察到导致神经系统症状,如共济失调和痉挛 [ 124 , 125 ]。这种罕见的遗传病的特点是影响负责激活 GP 的磷酸化酶的突变。尽管这种疾病没有证据表明镉参与了这一过程,但它确实表明 GP 下调足以产生神经毒性作用。其他神经退行性疾病,例如庞贝病,也与糖原积累有关[ 126 , 127 ]。
7.2. Evidence Contradicting the Original Theory Regarding Cadmium’s Functional Impairment of bGP
7.2.与镉损害 bGP 功能的原始理论相矛盾的证据
虽然确实存在证据支持镉与糖原相关的神经毒性的最初假设,但也存在反对这一理论的证据。 Roelfzema 研究表明,接触镉后糖原积累增加,但矛盾的是,镉诱导 GP 活性增加,这表明虽然镉可能导致糖原积累增加,但它可能以不涉及 GP 糖原的机制实现。磷酸化酶抑制[ 122 ]。此外,动物研究探讨了镉暴露对糖原的影响,这些研究在很大程度上发现糖原消耗而不是积累可能是镉糖原相关毒性的主要原因。例如,在攀鲈中,镉暴露导致肌肉和肝脏组织中的糖原水平显着降低,这表明镉可能通过抑制糖原合成来影响储存糖原的能力[ 128 ]。在淡水双壳贝类中,暴露于 CdCl 2 (水中 7.0 ppm 和 12.0 ppm)会增加其腹足动物器官中糖原的降解,从而增加该物种的能量储存消耗率[ 87 ]。此外,在大鼠中,浓度为 2.6 和 5.2 mg/kg 体重的 CdCl 2被发现会减少肝脏中的糖原储备,这表明哺乳动物物种中的糖原储存也受到影响 [ 129 ]。
关于镉诱导糖原消耗的机制明显缺乏假设。镉可能减少糖原储备的两个一般原因包括(1)通过干扰参与糖原合成的酶来损害糖原生成,或(2)过度激活糖原分解,这可以通过降低抑制剂的浓度来实现糖原分解作用,例如胰岛素。有趣的是,人们发现镉可以减少胰岛素的释放,这可能有助于解释在没有胰岛素抑制剂的情况下,由于糖原分解过度而导致糖原储备减少的数据[ 130 ]。此外,镉干扰 PI3 激酶/Akt/mTOR 信号传导,从而下调 FOXO1(一种刺激糖原分解的转录因子)和糖原合成酶激酶 3β(一种通过抑制/磷酸化促进糖原分解的酶),从而导致糖原分解过度激活。糖原合成酶 [ 131 , 132 ]。受镉影响的 Akt/糖原合酶激酶 3β 信号传导也与神经元细胞凋亡有关 [ 102 ]。另一种理论简单地指出,处于应激条件下的细胞往往需要更多的能量来解决应激源,从而增加糖原分解以促进葡萄糖储存中的 ATP 生成 [ 76 , 133 ]。真正的机制可能相当复杂,涉及多种酶和细胞过程。
7.3. Human Data Pointing towards Cadmium as a Glycogen-Disruptor
7.3.人类数据表明镉是一种糖原干扰物
人类流行病学数据表明,镉暴露是导致糖原代谢失调的一个因素。当糖原代谢因接触重金属(包括镉)而受到损害时,观察到的代谢综合征的发生率较高[ 134 ]。此外,镉暴露还会影响糖原/胰岛素途径,从而增加患糖尿病的风险,从而导致糖尿病神经病变等症状[ 130,135,136 ]。
神经退行性疾病可能是由于长期糖原代谢失调引起的。这在 AD [ 137 , 138 ] 和拉福拉病等遗传性疾病 [ 139 ] 中尤其明显。在 AD 病理学中,由于镉毒性而上调的糖原合酶激酶 3β 被认为是一种 tau 激酶,有助于神经退行性疾病的进展 [ 140 ]。此外,糖原代谢功能障碍与精神分裂症有关,这表明镉由于其自身的代谢功能障碍诱导特性也可能导致精神分裂症等神经精神疾病[ 141 ]。
7.4. Next Steps for Resolving Cadmium’s Effects on Neuronal Glycogen
7.4.解决镉对神经元糖原影响的后续步骤
总体而言,考虑到支持和反对 bGP 功能损伤导致糖原积累这一最初假设的证据,镉似乎可能会干扰糖原合成和分解。糖原储备的精确结果,即它们是增加还是减少,取决于所研究的特定物种和感兴趣的组织。目前尚不清楚这些机制(糖原耗尽或糖原过多)在多大程度上与人类镉神经毒性最具有临床和生物学相关性。然而,从人类临床数据可以清楚地看出,镉暴露会深刻影响人类健康和疾病,其方式往往涉及异常的糖原代谢。未来必须进行的研究来解决这些差异,包括直接研究镉暴露后神经胶质细胞中糖原储备的神经组织实验,分析糖原生成和糖原分解途径中所有酶对镉暴露的反应的生物活性(即,不只是糖原磷酸化酶),并在镉中毒后检查人体组织学样本(如果有)。
8. Possible Protective Measures against Cadmium Neurotoxicity
8. 针对镉神经毒性的可能保护措施
镉长期以来被认为是氧化应激的有效诱导剂,破坏神经系统内活性氧和抗氧化剂之间的平衡,从而对神经健康构成重大威胁。本综述的前面部分阐明了镉发挥神经毒性作用的复杂机制,从线粒体功能障碍到胆碱能神经元损失。然而,一个有希望的研究途径已经出现,揭示了抗氧化剂对镉引起的神经毒性的潜在保护措施。
含硫醇的蛋白质,包括谷胱甘肽 (GSH)、牛血清白蛋白 (BSA) 和硒蛋白 P,也已成为减轻镉引起的神经毒性的关键因素。这些蛋白质以其清除自由基和与镉结合的能力而闻名,可以作为抵御镉对神经组织有害影响的保护盾[ 36 ]。通过减少游离镉与线粒体蛋白上关键硫醇基团结合的可用性,这些抗氧化剂有助于维持线粒体功能并防止镉诱导的通透性转变孔 (PTP) 打开,最终保持神经元的完整性。
有实验证据表明特定抗氧化剂可减轻镉引起的神经毒性。布兰卡等人。证明抗氧化剂对镉具有一定的对抗能力,因为将抗氧化剂 α-生育酚乙酸酯应用于暴露于镉的大鼠脑内皮细胞可防止 GRP78 的上调,GRP78 是 ER 应激的标志物,负责 BBB 结构的下游损伤。 α-生育酚乙酸酯可防止 GRP78 上调,这表明抗氧化剂具有针对氧化剂依赖性 ER 应激反应的保护能力,这种应激反应最终会破坏 BBB 结构并导致镉诱导的神经毒性 [ 100 ]。
在一项利用斯普拉格-道利大鼠的研究中,镉暴露导致睾丸内的氧化应激和自噬。然而,补充抗氧化剂槲皮素显示出对抗镉引起的睾丸损伤的保护能力[ 142 ]。这一发现凸显了抗氧化剂减轻镉对神经组织不利影响的潜力。此外,槲皮素在CdCl 2诱导的雄性大鼠海马神经毒性中的神经保护潜力表明,槲皮素通过增强CdCl 2处理的大鼠的记忆功能和减轻海马损伤而发挥有益的影响。槲皮素可提高谷胱甘肽 (GSH) 和锰超氧化物歧化酶 (MnSOD) 等抗氧化剂的水平。此外,槲皮素上调SIRT1(一种参与细胞应激反应的蛋白质)的活性,抑制AChE的活性,抑制ROS的产生,并增加脑源性神经营养因子(BDNF)的水平,脑源性神经营养因子(BDNF)是一种对神经元存活和功能至关重要的蛋白质[ 143 ]。
另一种值得注意的抗氧化剂,β-胡萝卜素,在保护神经元健康免受镉引起的毒性的侵袭方面显示出良好的前景。在一项针对大鼠的综合研究中,镉暴露导致脂质过氧化(LPO)显着增加,表明神经组织内发生氧化损伤。镉暴露还与血清尿素和血尿素氮水平升高有关,表明肾功能障碍。 β-胡萝卜素预处理可改善镉引起的 LPO 水平、血清尿素和血尿素氮水平升高,强调其在对抗镉引起的氧化应激和肾脏健康方面的作用[ 144 ]。
在镉清除方面,乙二胺四乙酸 (EDTA) 已显示出有希望的结果。 EDTA 作为螯合剂广泛用于螯合二价和三价金属离子。 EDTA 通过四个羧酸盐和两个胺基与金属结合,并与 Mn (II)、Cu (II)、Fe (III) 和 Co (III) 形成特别牢固的键 [ 145 ]。由于这种特性,EDTA 被用作去除铅和镉以减轻金属毒性的药物[ 146 ]。 Waters 等人 (2001) 等研究显示 EDTA 螯合疗法具有有益效果,观察到尿液中镉的流失量显着增加 [ 147 ]。虽然这些研究的重点是镉从体内的流失,但 Fulgenzi 等人最近的研究(2020)已深入研究了镉的神经毒性方面。 EDTA 在有毒金属螯合疗法中的使用已被证明对神经退行性疾病具有有益作用,这在未来针对镉的保护措施方面显示出有希望的结果[ 148 ]。
最近有证据表明镉诱导的神经毒性与 ROS 增加和线粒体依赖性 ER 应激有关,Mostafa 等人于 2019 年研究了水合芦丁 (RH) 的作用,这是一种众所周知的神经保护物质的抗氧化剂黄酮类化合物 [ 149 ]。结果表明,RH抑制线粒体通透性转换孔,增强线粒体偶联,并抑制大脑中线粒体细胞色素c的释放。此外,RH 抑制线粒体 Bax 易位,并且如前所述,Kim 等人,2013 年证明 Bax 表达诱导细胞凋亡,支持 RH 可能是针对镉诱导的神经毒性的潜在保护措施 [ 47 ]。
半胱氨酸还被观察到可以逆转镉引起的骨骼神经肌肉神经传递阻断[ 25 ]。暴露于 100 µM 镉的小鸡双腹颈神经肌肉制剂在暴露后约 20 分钟内表现出抽搐高度降低 75%,并且应用 1 mM 半胱氨酸能够完全逆转神经肌肉接头处的这种阻断。作者还对暴露于不同浓度镉(10-100 µM)的小鼠膈神经肌肉制剂的运动神经末梢的神经周围波形进行了细胞外记录,并报告镉阻断了与钙电流相关的长期正向偏转。这种镉诱导的阻断可被 300 µM 半胱氨酸部分逆转,并被 1 mM 半胱氨酸完全逆转。半胱氨酸可以通过螯合镉来逆转镉诱导的钙电流阻断,因为据报道,镉可以与金属硫蛋白和关键抗氧化剂谷胱甘肽的半胱氨酸残基上的硫醇基团结合[ 150 ]。在后来的综述中,Braga 和 Rowan 指出,半胱氨酸可以阻断镉的所有细胞外效应,强调开发半胱氨酸疗法以减轻镉神经毒性的重要性[ 25 ]。包含外源性螯合剂和抗氧化剂的联合疗法已用于治疗镉毒性,但在治疗和预防镉神经毒性的最佳实践方面仍有许多待探索[ 151 ]。进一步的研究应集中于镉、半胱氨酸和其他螯合剂在神经系统中的相互作用,以开发有效的疗法来对抗镉引起的急性和慢性毒性暴露的神经毒性。
锌也被建议作为对抗镉神经毒性的保护剂。口服锌补充剂被认为可以预防与镉诱导的氧化应激相关的自由基,并减轻镉诱导的肾毒性[ 151 ]。口服补充剂也被证明可以通过减少小鼠模型中 Aβ 斑块的形成来减缓与镉中毒和阿尔茨海默氏病相关的神经元衰老的进展 [ 152 ]。然而,锌对神经毒性的保护功能要复杂得多,并且似乎具有剂量依赖性。低剂量的锌补充剂可以保护大鼠海马神经元免受镉引起的神经传递破坏,但高剂量的锌补充会增强镉的神经毒性[ 32 ]。需要进行更多研究来阐明镉、锌、阿尔茨海默病和氧化应激之间的复杂关系,以开发针对镉神经毒性的锌基疗法。
9. Conclusions 9. 结论
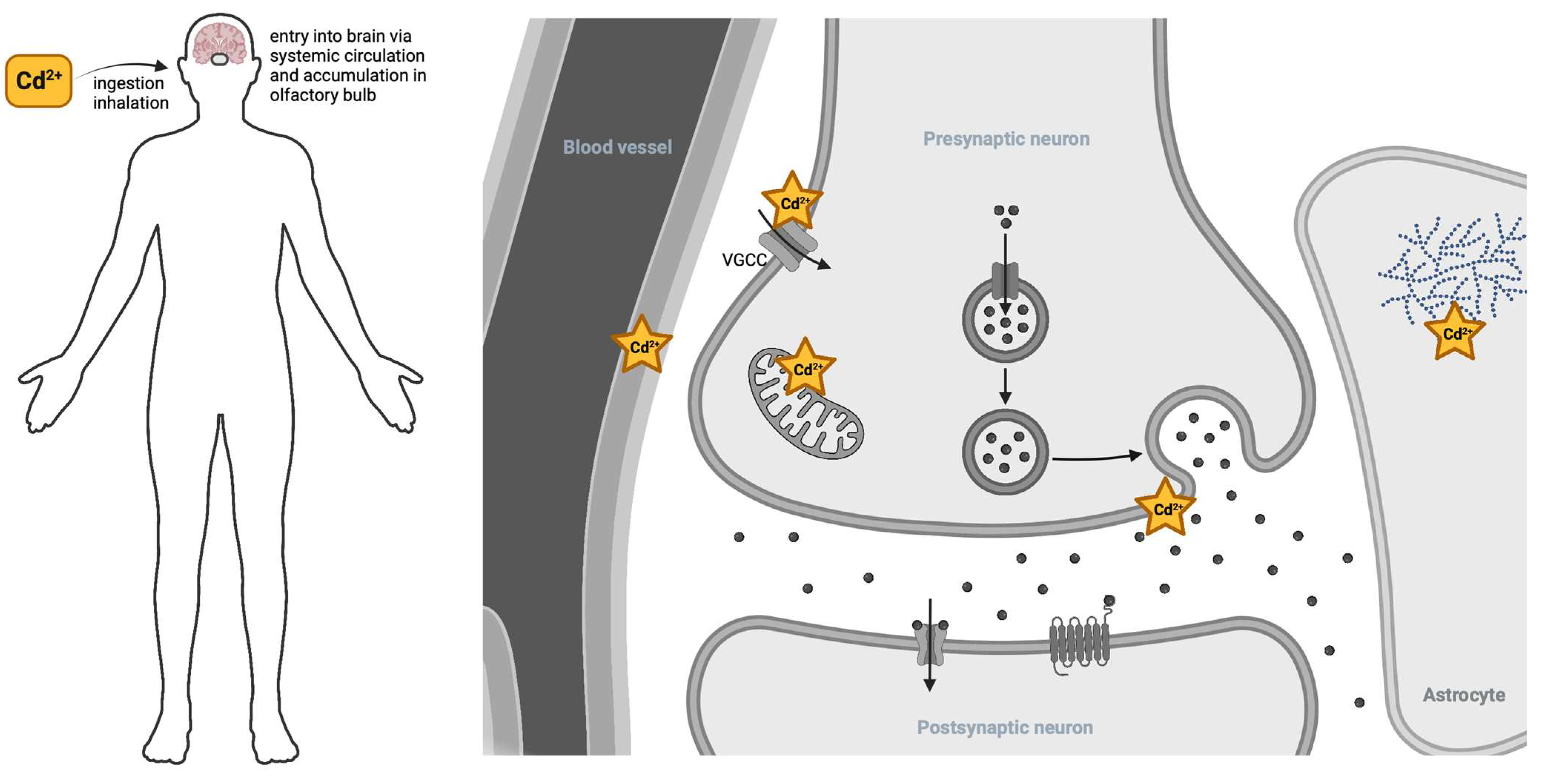
镉对神经系统有几种主要的毒性损害途径(图 4 )。镉首先通过吸入进入人体,从而积聚在嗅球中,或者更常见的是通过摄入,最终通过血脑屏障进入血液和中枢神经系统。镉会削弱血脑屏障,从而增加镉进一步进入的机会。然后镉可以通过钙和锌转运蛋白进入细胞和细胞区室。细胞内镉可以严重破坏糖原代谢,改变神经递质信号传导,并破坏线粒体功能,从而增加神经退行性结果的风险。

图 4.镉进入神经系统(左)。镉通过吸入或摄入进入神经系统。常见来源是在受镉污染的土壤中种植的食品。在较小程度上,吸入工业来源的镉烟雾或香烟烟雾可以通过肺部和嗅球进入。镉在大脑内的作用部位(右)。一旦进入循环,镉就会降低血脑屏障的完整性,从而进一步渗透到神经组织中。镉通过阳离子渗透的整合膜蛋白(例如电压门控钙通道(VGCC))进入细胞。一旦进入星形胶质细胞的细胞质,镉就会破坏糖原的利用。在神经元内部,镉通过囊泡循环扰乱线粒体功能和神经递质的胞吐作用。
镉神经毒性的几个领域值得进一步研究。在所有情况下,必须更多地关注这些研究的镉浓度,因为当前的研究代表了广泛的浓度,其中一些浓度比人类平均镉负荷高出几个数量级。首先,需要更多的研究来描述镉、PTP 和弱化 ΔΨm 之间的分子关系。必须进行进一步的研究来阐明镉与糖生成和糖原分解的复杂相互作用。尽管目前的研究表明镉与神经退行性疾病(尤其是阿尔茨海默病)之间存在初步联系,但未来的研究应努力消除这种联系。特别是,关于镉通过破坏锌稳态而在神经元衰老发展中的作用的进一步研究与神经退行性疾病的病因学直接相关。镉暴露和神经退行性疾病共有的生物标志物的鉴定可能有助于开发神经退行性疾病改善和预防的靶向疗法。研究还可能有助于开发减轻神经毒性的保护措施和疗法,例如抗氧化剂和螯合剂联合疗法以及口服锌补充剂。
随着全球工业化持续上升,镉暴露的发生率预计将增加。医生和公共卫生官员必须意识到镉的神经毒性作用,事实证明镉具有累积性和强毒性。
了解镉神经毒性的进展将促进对神经毒性本身以及密切相关的、日益常见的神经退行性疾病的理解和治疗开发。
Author Contributions 作者贡献
概念化,RCB;写作——初稿、MAA、YML 和 CTH;写作——审查和编辑、MAA、YML、CTH 和 RCB;可视化、CTH 和 RCB;监督、RCB;项目管理,RCB 所有作者均已阅读并同意稿件的出版版本。
Funding 资金
这项研究没有获得外部资助。
Institutional Review Board Statement
机构审查委员会声明
Informed Consent Statement
知情同意书
Data Availability Statement
数据可用性声明
Acknowledgments 致谢
作者要感谢圣母大学环境暴露和神经系统课程的本科生对本文的支持。作者感谢 Perry Harlan Cliburn 对本综述的宝贵评论。作者感谢圣母科学学院使用 BioRender 2023,这是用于本手稿中所有图形的图形制作软件。
Conflicts of Interest 利益冲突
作者没有需要声明的利益冲突。
References 参考
- Substance Priority List|ATSDR. Available online: https://www.atsdr.cdc.gov/spl/index.html (accessed on 10 October 2023).
物质优先列表|ATSDR。在线提供: https://www.atsdr.cdc.gov/spl/index.html (2023 年 10 月 10 日访问)。 - Public Health Statement Cadmium. Agency for Toxic Substances and Disease Registry. 2012. Available online: https://www.atsdr.cdc.gov/ToxProfiles/tp5-c1-b.pdf (accessed on 10 October 2023).
公共卫生声明镉。有毒物质和疾病登记局。 2012 年。在线提供: https://www.atsdr.cdc.gov/ToxProfiles/tp5-c1-b.pdf (2023 年 10 月 10 日访问)。 - Klaassen, C.; Watkins, J.B., III. Casarett and Doull’s Essentials of Toxicology, 1st ed.; McGraw-Hill Professional: New York, NY, USA, 2003. [Google Scholar]
克拉森,C.;沃特金斯,JB,III。卡萨雷特和杜尔的《毒理学要点》 ,第一版;麦格劳-希尔专业人士:美国纽约州纽约市,2003 年。[ Google 学术搜索] - The Facts on Cadmium. Available online: https://sites.dartmouth.edu/toxmetal/more-metals/cadmium-an-illusive-presence/the-facts-on-cadmium/ (accessed on 1 October 2023).
关于镉的事实。在线提供: https://sites.dartmouth.edu/toxmetal/more-metals/cadmium-an-illusive-presence/the-facts-on-cadmium/ (2023 年 10 月 1 日访问)。 - Mena, C.; Cabrera, C.; Lorenzo, M.L.; López, M.C. Cadmium levels in wine, beer and other alcoholic beverages: Possible sources of contamination. Sci. Total Environ. 1996, 181, 201. [Google Scholar] [CrossRef] [PubMed]
梅纳,C.;卡布雷拉,C.;洛伦佐,ML; López, MC 葡萄酒、啤酒和其他酒精饮料中的镉含量:可能的污染源。科学。整体环境。 1996 , 181 , 201. [谷歌学术] [交叉引用] [ PubMed ] - Satarug, S.; Moore, M.R. Adverse Health Effects of Chronic Exposure to Low-Level Cadmium in Foodstuffs and Cigarette Smoke. Environ. Health Perspect. 2004, 112, 1099–1103. [Google Scholar] [CrossRef] [PubMed]
萨塔鲁格,S.; Moore, MR 长期接触食品和香烟烟雾中的低含量镉对健康的不利影响。环境。健康视角。 2004 , 112 , 1099–1103。 [谷歌学术] [交叉引用] [ PubMed ] - Sunderman, F.W. Nasal toxicity, carcinogenicity, and olfactory uptake of metals. Ann. Clin. Lab. Sci. 2001, 31, 3–24. [Google Scholar]
Sunderman,FW 金属的鼻毒性、致癌性和嗅觉吸收。安.临床。实验室。科学。 2001 , 31 , 3–24。 [谷歌学术] - Suwazono, Y.; Kido, T.; Nakagawa, H.; Nishijo, M.; Honda, R.; Kobayashi, E.; Dochi, M.; Nogawa, K. Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 2009, 14, 77–81. [Google Scholar] [CrossRef]
诹访园,Y.;基多,T.;中川,H.;西条,M.;本田,R。小林,E.;多奇,M.;野川,K。停止接触镉后居民尿液中镉的生物半衰期。生物标志物2009 , 14 , 77–81。 [谷歌学术] [交叉引用] - International Agency for Research on Cancer. Beryllium, Cadmium, Mercury, and Exposures in the Glass Manufacturing Industry; International Agency for Research on Cancer: Lyon, France, 1993; Volume 58, pp. 1–415. [Google Scholar]
国际癌症研究机构。铍、镉、汞和玻璃制造业中的暴露;国际癌症研究机构:法国里昂,1993 年;第 58 卷,第 1–415 页。 [谷歌学术] - What Is the Biological Fate of Cadmium in the Body? Available online: https://www.atsdr.cdc.gov/csem/cadmium/Biological-Fate.html#:~:text=Cadmium%20is%20transported%20in%20the,the%20major%20mechanism%20of%20elimination (accessed on 12 October 2023).
镉在体内的生物归宿是什么?在线提供: https://www.atsdr.cdc.gov/csem/cadmium/Biological-Fate.html# :~:text=Cadmium%20is%20transported%20in%20the,the%20major%20mechanism%20of%20elimination (访问日期:2023 年 10 月 12 日)。 - Vinceti, M.; Filippini, T.; Mandrioli, J.; Violi, F.; Bargellini, A.; Weuve, J.; Fini, N.; Grill, P.; Michalke, B. Lead, cadmium and mercury in cerebrospinal fluid and risk of amyotrophic lateral sclerosis: A case-control study. J. Trace Elem. Med. Biol. 2017, 43, 121–125. [Google Scholar] [CrossRef]
文塞蒂,M.;菲利皮尼,T.;曼德里奥利,J.;维奥利,F.;巴杰利尼,A.;韦夫,J.;菲尼,N.;格里尔,P.; Michalke, B. 脑脊液中的铅、镉和汞与肌萎缩侧索硬化症的风险:一项病例对照研究。 J.特雷斯·埃莱姆。医学。生物。 2017 , 43 , 121–125。 [谷歌学术] [交叉引用] - Ma, Y.; Su, Q.; Yue, C.; Zou, H.; Zhu, J.; Zhao, H.; Song, R.; Liu, Z. The Effect of Oxidative Stress-Induced Autophagy by Cadmium Exposure in Kidney, Liver, and Bone Damage, and Neurotoxicity. Int. J. Mol. Sci. 2022, 23, 13491. [Google Scholar] [CrossRef]
可能。;苏,Q。岳,C.;邹,H.;朱,J。赵,H。宋,R。 Liu, Z. 镉暴露对氧化应激诱导的自噬对肾脏、肝脏和骨损伤以及神经毒性的影响。国际。 J.莫尔。科学。 2022 , 23 , 13491. [谷歌学术] [交叉引用] - Viaene, M.K.; Masschelein, R.; Leenders, J.; De Groof, M.; Swerts, L.J.V.C.; Roels, H.A. Neurobehavioural effects of occupational exposure to cadmium: A cross sectional epidemiological study. Occup. Environ. Med. 2000, 57, 19–27. [Google Scholar] [CrossRef]
维埃内,MK;马舍莱因,R.;利德斯,J.;德格鲁夫,M.; Swerts,LJVC; Roels,HA 职业接触镉的神经行为影响:一项横断面流行病学研究。占领。环境。医学。 2000 , 57 , 19–27。 [谷歌学术] [交叉引用] - Li, H.; Wang, Z.; Fu, Z.; Yan, M.; Wu, N.; Wu, H.; Yin, P. Associations between blood cadmium levels and cognitive function in a cross-sectional study of US adults aged 60 years or older. BMJ Open 2018, 8, e020533. [Google Scholar] [CrossRef]
李,H。王,Z。傅,Z。严,M。吴,N。吴,H。 Yin, P. 对美国 60 岁或以上成年人进行的一项横断面研究中血镉水平与认知功能之间的关联。 BMJ 公开赛2018,8 , e020533 。 [谷歌学术] [交叉引用] - Ciesielski, T.; Weuve, J.; Bellinger, D.C.; Schwartz, J.; Lanphear, B.; Wright, R.O. Cadmium Exposure and Neurodevelopmental Outcomes in U.S. Children. Environ. Health Perspect. 2012, 120, 758–763. [Google Scholar] [CrossRef]
西谢尔斯基,T.;韦夫,J.;贝林格,华盛顿特区;施瓦茨,J.;兰菲尔,B.; Wright,RO 美国儿童的镉暴露和神经发育结果。环境。健康视角。 2012 , 120 , 758–763。 [谷歌学术] [交叉引用] - Kippler, M.; Tofail, F.; Hamadani, J.D.; Gardner, R.M.; Grantham-McGregor, S.M.; Bottai, M.; Vahter, M. Early-life cadmium exposure and child development in 5-year-old girls and boys: A cohort study in rural bangladesh. Environ. Health Perspect. 2012, 120, 1462–1468. [Google Scholar] [CrossRef]
基普勒,M.;托法尔,F.;哈马达尼,法学博士;加德纳,RM;格兰瑟姆·麦格雷戈,SM;博泰,M.; Vahter, M. 5 岁女孩和男孩的早期镉暴露与儿童发育:孟加拉国农村地区的一项队列研究。环境。健康视角。 2012 , 120 , 1462–1468。 [谷歌学术] [交叉引用] - Forcella, M.; Lau, P.; Oldani, M.; Melchioretto, P.; Bogni, A.; Gribaldo, L.; Fusi, P.; Urani, C. Neuronal specific and non-specific responses to cadmium possibly involved in neurodegeneration: A toxicogenomics study in a human neuronal cell model. Neurotoxicology 2020, 76, 162–173. [Google Scholar] [CrossRef] [PubMed]
福塞拉,M.;刘,P。奥尔达尼,M.;梅尔基奥雷托,P.;博尼,A.;格里巴尔多,L.;富西,P.; Urani, C. 神经元对镉的特异性和非特异性反应可能与神经变性有关:人类神经元细胞模型的毒物基因组学研究。神经毒理学2020 , 76 , 162–173。 [谷歌学术] [交叉引用] [ PubMed ] - Raj, K.; Kaur, P.; Gupta, G.D.; Singh, S. Metals associated neurodegeneration in Parkinson’s disease: Insight to physiological, pathological mechanisms and management. Neurosci. Lett. 2021, 753, 135873. [Google Scholar] [CrossRef] [PubMed]
拉杰,K.;考尔,P.;古普塔,GD; Singh, S. 帕金森病中金属相关的神经变性:对生理、病理机制和管理的洞察。神经科学。莱特。 2021 , 753 , 135873.[谷歌学术][交叉引用][ PubMed ] - Min, J.; Min, K. Blood cadmium levels and Alzheimer’s disease mortality risk in older US adults. Environ. Health 2016, 15, 69. [Google Scholar] [CrossRef]
敏,J.; Min, K. 美国老年人的血液镉水平和阿尔茨海默病死亡风险。环境。健康2016 , 15 , 69. [谷歌学术] [交叉引用] - Branca, J.J.V.; Morucci, G.; Pacini, A. Cadmium-induced neurotoxicity: Still much ado. Neural Regen. Res. 2018, 13, 1879–1882. [Google Scholar] [PubMed]
布兰卡,JJV;莫鲁奇,G.; Pacini, A. 镉引起的神经毒性:仍然很麻烦。神经再生。资源。 2018 , 13 , 1879–1882。 [谷歌学术] [考研] - Witkowska, D.; Słowik, J.; Chilicka, K. Heavy Metals and Human Health: Possible Exposure Pathways and the Competition for Protein Binding Sites. Molecules 2021, 26, 6060. [Google Scholar] [CrossRef]
维特科夫斯卡,D.;斯沃维克,J.; Chilicka, K. 重金属与人类健康:可能的暴露途径和蛋白质结合位点的竞争。分子2021 , 26 , 6060. [谷歌学术] [交叉引用] - Tjälve, H.; Henriksson, J.; Tallkvist, J.; Larsson, B.S.; Lindquist, N.G. Uptake of Manganese and Cadmium from the Nasal Mucosa into the Central Nervous System via Olfactory Pathways in Rats. Pharmacol. Toxicol. 1996, 79, 347–356. [Google Scholar] [CrossRef]
Tjälve,H.;亨里克森,J.;塔尔克维斯特,J.;拉尔森,BS; Lindquist,NG 大鼠通过嗅觉途径将锰和镉从鼻粘膜吸收到中枢神经系统。药理学。毒性。 1996 , 79 , 347–356。 [谷歌学术] [交叉引用] - Choong, G.; Liu, Y.; Templeton, D.M. Interplay of calcium and cadmium in mediating cadmium toxicity. Chem.-Biol. Interact. 2014, 211, 54–65. [Google Scholar] [CrossRef]
钟,G.;刘,Y。 Templeton,DM 钙和镉在介导镉毒性中的相互作用。化学-生物。相互影响。 2014 , 211 , 54–65。 [谷歌学术] [交叉引用] - Usai, C.; Barberis, A.; Moccagatta, L.; Marchetti, C. Pathways of Cadmium Influx in Mammalian Neurons. J. Neurochem. 1999, 72, 2154–2161. [Google Scholar] [CrossRef]
乌萨伊,C.;巴伯里斯,A.;莫卡加塔,L.; Marchetti, C. 哺乳动物神经元中镉流入的途径。 J.神经化学。 1999 , 72 , 2154–2161。 [谷歌学术] [交叉引用] - Braga, M.F.M.; Rowan, E.G. Reversal by cysteine of the cadmium-induced block of skeletal neuromuscular transmission in vitro. Br. J. Pharmacol. 1992, 107, 95–100. [Google Scholar] [CrossRef]
布拉加,MFM; Rowan,EG 半胱氨酸逆转镉诱导的体外骨骼神经肌肉传递阻断。 Br。 J.Pharmacol。 1992 , 107 , 95–100。 [谷歌学术] [交叉引用] - Minami, A.; Takeda, A.; Nishibaba, D.; Takefuta, S.; Oku, N. Cadmium toxicity in synaptic neurotransmission in the brain. Brain Res. 2001, 894, 336. [Google Scholar] [CrossRef]
南,A.;武田,A.;西马巴,D.;武田,S.; Oku, N. 大脑突触神经传递中的镉毒性。脑研究。 2001 , 894 , 336. [谷歌学术] [交叉引用] - Thévenod, F.; Schulz, I. H+-dependent calcium uptake into an IP3-sensitive calcium pool from rat parotid gland. Am. J. Physiol.-Gastrointest. Liver Physiol. 1988, 255, 429–440. [Google Scholar] [CrossRef] [PubMed]
特维诺,F.; Schulz,I. H +依赖性钙从大鼠腮腺摄取到 IP3 敏感钙池中。是。 J.生理学-胃肠测试。肝脏生理学。 1988 , 255 , 429–440。 [谷歌学术] [交叉引用] [ PubMed ] - Tsentsevitsky, A.; Petrov, A. Synaptic mechanisms of cadmium neurotoxicity. Neural Regen. Res. 2021, 16, 1762–1763. [Google Scholar] [PubMed]
岑采维茨基,A.; Petrov, A. 镉神经毒性的突触机制。神经再生。差距。 2021 , 16 , 1762–1763。 [谷歌学术] [考研] - Mimouna, S.B.; Chemek, M.; Boughammoura, S.; Banni, M.; Messaoudi, I. Early-Life Exposure to Cadmium Triggers Distinct Zn-Dependent Protein Expression Patterns and Impairs Brain Development. Biol. Trace Elem. Res. 2018, 184, 409–421. [Google Scholar] [CrossRef]
米穆纳,SB;切梅克,M.;布哈姆莫拉,S.;班尼,M.; Messaoudi, I. 生命早期接触镉会触发独特的锌依赖性蛋白质表达模式并损害大脑发育。生物。跟踪埃莱姆。资源。 2018 , 184 , 409–421。 [谷歌学术] [交叉引用] - McAllister, B.B.; Dyck, R.H. Zinc transporter 3 (ZnT3) and vesicular zinc in central nervous system function. Neurosci. Biobehav. Rev. 2017, 80, 329–350. [Google Scholar] [CrossRef] [PubMed]
麦卡利斯特,BB; Dyck,RH 锌转运蛋白 3 (ZnT3) 和囊泡锌在中枢神经系统中的功能。神经科学。生物行为。 2017 年修订版, 80,329–350 。 [谷歌学术] [交叉引用] [ PubMed ] - Xu, Y.; Xiao, G.; Liu, L.; Lang, M. Zinc transporters in Alzheimer’s disease. Mol. Brain 2019, 12, 106. [Google Scholar] [CrossRef]
徐,Y。肖,G。刘,L。 Lang, M. 阿尔茨海默病中的锌转运蛋白。摩尔。大脑2019 , 12 , 106. [谷歌学术] [交叉引用] - Ben Mimouna, S.; Le Charpentier, T.; Lebon, S.; Van Steenwinckel, J.; Messaoudi, I.I.; Gressens, P. Involvement of the synapse-specific zinc transporter ZnT3 in cadmium-induced hippocampal neurotoxicity. J. Cell. Physiol. 2019, 234, 15872. [Google Scholar] [CrossRef]
本·米莫纳,S.;勒·查庞蒂埃,T.;莱邦,S.;范·斯廷温克尔,J.;迈萨乌迪,II; Gressens, P. 突触特异性锌转运蛋白 ZnT3 参与镉诱导的海马神经毒性。 J.细胞。生理学。 2019 , 234 , 15872. [谷歌学术] [交叉引用] - Rolfe, D.F.; Brown, G.C. Cellular energy utilization and molecular origin of standard metabolic rate in mammals. Physiol. Rev. 1997, 77, 731–758. [Google Scholar] [CrossRef]
罗尔夫,DF; Brown,GC 哺乳动物细胞能量利用和标准代谢率的分子起源。生理学。修订版1997 , 77 , 731–758。 [谷歌学术] [交叉引用] - Brand, M.D.; Orr, A.L.; Perevoshchikova, I.V.; Quinlan, C.L. The role of mitochondrial function and cellular bioenergetics in ageing and disease. Br. J. Dermatol. 2013, 169 (Suppl. S2), 1–8. [Google Scholar] [CrossRef]
品牌,医学博士;奥尔,阿拉巴马州;佩列沃什奇科娃,IV; Quinlan, CL 线粒体功能和细胞生物能量学在衰老和疾病中的作用。 Br。 J.德马托尔。 2013,169 (补编 S2), 1-8 。 [谷歌学术] [交叉引用] - Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef]
佐罗娃,LD;波普科夫,弗吉尼亚州;普洛特尼科夫,EY;西拉切夫,DN;佩夫兹纳,IB;扬考斯卡斯,SS;弗吉尼亚州巴边科;佐罗夫,SD;巴拉基列娃,AV;尤哈佐娃,M.;等人。线粒体膜电位。肛门。生物化学。 2018 , 552 , 50–59。 [谷歌学术] [交叉引用] - Belyaeva, E.; Glazunov, V.; Korotkov, S. Cd2-promoted mitochondrial permeability transition: A comparison with other heavy metals. Acta Biochim. Pol. 2004, 51, 545–551. [Google Scholar] [CrossRef]
别利亚耶娃,E.;格拉祖诺夫,V.; Korotkov, S. Cd2 促进的线粒体通透性转变:与其他重金属的比较。生物化学学报。波尔。 2004 , 51 , 545–551。 [谷歌学术] [交叉引用] - Branca, J.J.V.; Pacini, A.; Gulisano, M.; Taddei, N.; Fiorillo, C.; Becatti, M. Cadmium-Induced Cytotoxicity: Effects on Mitochondrial Electron Transport Chain. Front. Cell Dev. Biol. 2020, 8, 604377. [Google Scholar] [CrossRef] [PubMed]
布兰卡,JJV;帕西尼,A.;古利萨诺,M.;塔代伊,N.;菲奥里洛,C.; Becatti, M. 镉诱导的细胞毒性:对线粒体电子传输链的影响。正面。细胞开发。生物。 2020 , 8 , 604377. [谷歌学术] [ CrossRef ] [ PubMed ] - Xu, S.; Pi, H.; Chen, Y.; Zhang, N.; Guo, P.; Lu, Y.; He, M.; Xie, J.; Zhong, M.; Zhang, Y.; et al. Cadmium induced Drp1-dependent mitochondrial fragmentation by disturbing calcium homeostasis in its hepatotoxicity. Cell Death Dis. 2013, 4, e540. [Google Scholar] [CrossRef] [PubMed]
徐,S。皮,H.;陈,Y。张,N。郭,P。卢,Y。他,M。谢,J.;钟,M。张,Y。等人。镉在其肝毒性中通过扰乱钙稳态来诱导 Drp1 依赖性线粒体断裂。细胞死亡疾病。 2013 年4 月,e540。 [谷歌学术] [交叉引用] [ PubMed ] - Weidemann, M.J.; Erdelt, H.; Klingenberg, M. Adenine Nucleotide Translocation of Mitochondria. Eur. J. Biochem. 1970, 16, 313–335. [Google Scholar] [CrossRef] [PubMed]
魏德曼,MJ;埃尔德尔特,H.; Klingenberg,M. 线粒体的腺嘌呤核苷酸易位。欧元。 J.生物化学。 1970 , 16 , 313–335。 [谷歌学术] [交叉引用] [ PubMed ] - Appleby, R.D.; Porteous, W.K.; Hughes, G.; James, A.M.; Shannon, D.; Wei, Y.; Murphy, M.P. Quantitation and origin of the mitochondrial membrane potential in human cells lacking mitochondrial DNA. Eur. J. Biochem. 1999, 262, 108–116. [Google Scholar] [CrossRef]
艾普比,RD;波蒂斯,WK;休斯,G.;詹姆斯,上午;香农,D.;魏,Y。 Murphy, MP 缺乏线粒体 DNA 的人类细胞中线粒体膜电位的定量和起源。欧元。 J.生物化学。 1999 , 262 , 108–116。 [谷歌学术] [交叉引用] - Li, M.; Xia, T.; Jiang, C.; Li, L.; Fu, J.; Zhou, Z. Cadmium directly induced the opening of membrane permeability pore of mitochondria which possibly involved in cadmium-triggered apoptosis. Toxicology 2003, 194, 19–33. [Google Scholar] [CrossRef]
李,M。夏,T。江,C.;李,L。傅,J。 Zhou, Z. 镉直接诱导线粒体膜通透孔的开放,可能参与镉引发的细胞凋亡。毒理学2003 , 194 , 19–33。 [谷歌学术] [交叉引用] - Bround, M.J.; Bers, D.M.; Molkentin, J.D. A 20/20 view of ANT function in mitochondrial biology and necrotic cell death. J. Mol. Cell Cardiol. 2020, 144, A3–A13. [Google Scholar] [CrossRef]
布朗德,MJ; Bers,DM; Molkentin,JD 对线粒体生物学和坏死细胞死亡中 ANT 功能的 20/20 观点。 J.莫尔。细胞心脏。 2020 , 144 , A3–A13。 [谷歌学术] [交叉引用] - Loeffler, M.; Kroemer, G. The mitochondrion in cell death control: Certainties and incognita. Exp. Cell Res. 2000, 256, 19–26. [Google Scholar] [CrossRef]
洛夫勒,M.; Kroemer,G.细胞死亡控制中的线粒体:确定性和未知。过期。细胞研究。 2000 , 256 , 19–26。 [谷歌学术] [交叉引用] - Morishima, N.; Nakanishi, K.; Takenouchi, H.; Shibata, T.; Yasuhiko, Y. An endoplasmic reticulum stress-specific caspase cascade in apoptosis: Cytochrome c-independent activation of caspase-9 by caspase-12. J. Biol. Chem. 2002, 277, 34287–34294. [Google Scholar] [CrossRef]
森岛,N.;中西,K.;竹内,H.;柴田,T.; Yasuhiko, Y. 细胞凋亡中的内质网应激特异性 caspase 级联反应:caspase-12 对 caspase-9 的细胞色素 c 独立激活。 J.Biol。化学。 2002 , 277 , 34287–34294。 [谷歌学术] [交叉引用] - Kim, S.; Cheon, H.; Kim, S.; Juhnn, Y.; Kim, Y. Cadmium induces neuronal cell death through reactive oxygen species activated by GADD153. BMC Cell Biol. 2013, 14, 4. [Google Scholar] [CrossRef]
金,S。 Cheon,H.;金,S。尤恩,Y.; Kim, Y. 镉通过 GADD153 激活的活性氧诱导神经元细胞死亡。 BMC 细胞生物学。 2013 , 14 , 4. [谷歌学术] [交叉引用] - Ji, Y.; Wang, H.; Zhao, X.; Wang, Q.; Zhang, C.; Zhang, Y.; Zhao, M.; Chen, Y.; Meng, X.; Xu, D. Crosstalk between endoplasmic reticulum stress and mitochondrial pathway mediates cadmium-induced germ cell apoptosis in testes. Toxicol. Sci. 2011, 124, 446–459. [Google Scholar] [CrossRef]
吉,Y。王,H。赵X。王Q。张,C.;张,Y。赵,M。陈,Y。孟X.; Xu,D.内质网应激和线粒体途径之间的串扰介导镉诱导的睾丸生殖细胞凋亡。毒性。科学。 2011 , 124 , 446–459。 [谷歌学术] [交叉引用] - Nakagawa, T.; Zhu, H.; Morishima, N.; Li, E.; Xu, J.; Yankner, B.A.; Yuan, J. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-β. Nature 2000, 403, 98–103. [Google Scholar] [CrossRef] [PubMed]
中川,T.;朱,H.;森岛,N.;说谎。;徐,J。扬克纳,文学学士; Yuan, J. Caspase-12 通过淀粉样蛋白-β 介导内质网特异性细胞凋亡和细胞毒性。自然2000 , 403 , 98–103。 [谷歌学术] [交叉引用] [ PubMed ] - Shati, A.A. Resveratrol protects against cadmium chloride-induced hippocampal neurotoxicity by inhibiting ER stress and GAAD 153 and activating sirtuin 1/AMPK/Akt. Environ. Toxicol. 2019, 34, 1340–1353. [Google Scholar] [CrossRef] [PubMed]
Shati, AA 白藜芦醇通过抑制 ER 应激和 GAAD 153 以及激活 Sirtuin 1/AMPK/Akt 来防止氯化镉诱导的海马神经毒性。环境。毒性。 2019 , 34 , 1340–1353。 [谷歌学术] [交叉引用] [ PubMed ] - Rapizzi, E.; Pinton, P.; Szabadkai, G.; Wieckowski, M.R.; Vandecasteele, G.; Baird, G.; Tuft, R.A.; Fogarty, K.E.; Rizzuto, R. Recombinant expression of the voltage-dependent anion channel enhances the transfer of Ca2+ microdomains to mitochondria. J. Cell Biol. 2002, 159, 613–624. [Google Scholar] [CrossRef] [PubMed]
拉皮齐,E.;平顿,P.;萨巴德凯,G.;维科夫斯基先生;范德卡斯泰尔,G.;贝尔德,G.;塔夫特,RA;福加蒂,KE; Rizzuto, R. 电压依赖性阴离子通道的重组表达增强了 Ca 2+微结构域向线粒体的转移。 J.细胞生物学。 2002 , 159 , 613–624。 [谷歌学术] [交叉引用] [ PubMed ] - Wang, T.; Zhu, Q.; Cao, B.; Cai, Y.; Wen, S.; Bian, J.; Zou, H.; Song, R.; Gu, J.; Liu, X. Ca2+ transfer via the ER-mitochondria tethering complex in neuronal cells contribute to cadmium-induced autophagy. Cell Biol. Toxicol. 2021, 38, 469–485. [Google Scholar] [CrossRef]
王,T。朱Q。曹,B.;蔡,Y。温,S。卞,J。邹,H.;宋,R。顾,J。 Liu, X. Ca 2+通过神经元细胞中的 ER-线粒体束缚复合物转移有助于镉诱导的自噬。细胞生物学。毒性。 2021 , 38 , 469–485。 [谷歌学术] [交叉引用] - Ghavami, S.; Shojaei, S.; Yeganeh, B.; Ande, S.R.; Jangamreddy, J.R.; Mehrpour, M.; Christoffersson, J.; Chaabane, W.; Moghadam, A.R.; Kashani, H.H.; et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog. Neurobiol. 2014, 112, 24–49. [Google Scholar] [CrossRef]
加瓦米,S.;舒贾伊,S.;耶加内,B.;安德,SR;詹加姆雷迪,JR;梅尔普尔,M.;克里斯托弗森,J.;查巴内,W.;阿肯色州莫哈达姆;卡沙尼,HH;等人。神经退行性疾病中的自噬和凋亡功能障碍。程序。神经生物学。 2014,112,24-49 。 [谷歌学术] [交叉引用] - Chatterjee, S.; Sarkar, S.; Bhattacharya, S. Toxic metals and autophagy. Chem. Res. Toxicol. 2014, 27, 1887–1900. [Google Scholar] [CrossRef]
查特吉,S.;萨卡,S.; Bhattacharya, S. 有毒金属和自噬。化学。资源。毒性。 2014 , 27 , 1887–1900。 [谷歌学术] [交叉引用] - Wang, T.; Wang, Q.; Song, R.; Zhang, Y.; Zhang, K.; Yuan, Y.; Bian, J.; Liu, X.; Gu, J.; Liu, Z. Autophagy plays a cytoprotective role during cadmium-induced oxidative damage in primary neuronal cultures. Biol. Trace Elem. Res. 2015, 168, 481–489. [Google Scholar] [CrossRef]
王,T。王Q。宋,R。张,Y。张,K。袁,Y。卞,J。刘X。顾,J。 Liu, Z. 在原代神经元培养物中镉诱导的氧化损伤过程中,自噬发挥着细胞保护作用。生物。跟踪埃莱姆。资源。 2015 , 168 , 481–489。 [谷歌学术] [交叉引用] - Chen, Y.; McMillan-Ward, E.; Kong, J.; Israels, S.J.; Gibson, S.B. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell Death Differ. 2008, 15, 171–182. [Google Scholar] [CrossRef]
陈,Y。麦克米伦-沃德,E.;孔,J。以色列,SJ; Gibson, SB 氧化应激诱导自噬性细胞死亡,与转化细胞和癌细胞的凋亡无关。细胞死亡不同。 2008 , 15 , 171–182。 [谷歌学术] [交叉引用] - Zhang, H.; Dong, X.; Zhao, R.; Zhang, R.; Xu, C.; Wang, X.; Liu, C.; Hu, X.; Huang, S.; Chen, L. Cadmium results in accumulation of autophagosomes-dependent apoptosis through activating Akt-impaired autophagic flux in neuronal cells. Cell Signal 2019, 55, 26–39. [Google Scholar] [CrossRef]
张,H。董X.;赵,R。张,R。徐,C.;王X;刘,C.;胡X.;黄,S。 Chen, L. 镉通过激活神经元细胞中 Akt 受损的自噬流,导致自噬体依赖性细胞凋亡的积累。细胞信号2019 , 55 , 26–39。 [谷歌学术] [交叉引用] - Zhang, L.; Xia, Q.; Zhou, Y.; Li, J. Endoplasmic reticulum stress and autophagy contribute to cadmium-induced cytotoxicity in retinal pigment epithelial cells. Toxicol. Lett. 2019, 311, 105–113. [Google Scholar] [CrossRef]
张L。夏Q。周,Y。 Li,J.内质网应激和自噬有助于镉诱导的视网膜色素上皮细胞的细胞毒性。毒性。莱特。 2019 , 311 , 105–113。 [谷歌学术] [交叉引用] - Xu, C.; Liu, C.; Liu, L.; Zhang, R.; Zhang, H.; Chen, S.; Luo, Y.; Chen, L.; Huang, S. Rapamycin prevents cadmium-induced neuronal cell death via targeting both mTORC1 and mTORC2 pathways. Neuropharmacology 2015, 97, 35–45. [Google Scholar] [CrossRef] [PubMed]
徐,C.;刘,C.;刘,L。张,R。张,H。陈,S。罗,Y。陈L.; Huang, S. Rapamycin 通过靶向 mTORC1 和 mTORC2 途径来防止镉诱导的神经元细胞死亡。神经药理学2015 , 97 , 35–45。 [谷歌学术] [交叉引用] [ PubMed ] - Xu, C.; Chen, S.; Xu, M.; Chen, X.; Wang, X.; Zhang, H.; Dong, X.; Zhang, R.; Chen, X.; Gao, W.; et al. Cadmium Impairs Autophagy Leading to Apoptosis by Ca2+-Dependent Activation of JNK Signaling Pathway in Neuronal Cells. Neurochem. Res. 2021, 46, 2033–2045. [Google Scholar] [CrossRef] [PubMed]
徐,C.;陈,S。徐,M。陈X.;王X;张,H。董X.;张,R。陈X.;高,W。等人。镉会损害自噬,通过 Ca 2+依赖性激活神经元细胞中的 JNK 信号通路导致细胞凋亡。神经化学。资源。 2021、46、2033–2045 。 [谷歌学术] [交叉引用] [ PubMed ] - Hu, T.; Wei, X.; Zhang, X.; Cheng, F.; Shuai, X.; Zhang, L.; Kang, L. Protective effect of Potentilla anserine polysaccharide (PAP) on hydrogen peroxide induced apoptosis in murine splenic lymphocytes. Carbohydr. Polym. 2010, 79, 356–361. [Google Scholar] [CrossRef]
胡,T。魏,X。张X;程,F。帅,X.;张L。 Kang,L.委陵菜多糖(PAP)对过氧化氢诱导的小鼠脾淋巴细胞凋亡的保护作用。碳水化合物。聚合物。 2010 , 79 , 356–361。 [谷歌学术] [交叉引用] - QIN, X.; LI, L.; Qi, L.V.; YU, B.; YANG, S.; Tao, H.E.; ZHANG, Y. Neuroprotection of n-butanol extract from roots of Potentilla anserina on hypoxic injury in primary hippocampal neurons. Chin. Herb. Med. 2012, 4, 195–200. [Google Scholar]
秦X。李,L。齐,LV;于,B.;杨,S。陶,何;张,Y。委陵菜根正丁醇提取物对原代海马神经元缺氧损伤的神经保护作用。下巴。草本植物。医学。 2012 , 4 , 195–200。 [谷歌学术] - Shuai, X.; Hu, T.; Zhang, X.; Cheng, F.; Chen, J. Inhibitory action of Potentilla anserine polysaccharide fraction on H2O2-induced apoptosis of murine splenic lymphocytes. Yao Xue Xue Bao = Acta Pharm. Sin. 2009, 44, 987–993. [Google Scholar]
帅,X.;胡,T。张X;程,F。 Chen, J. 委陵菜多糖组分对 H2O2 诱导的小鼠脾淋巴细胞凋亡的抑制作用。药学学报 = Acta Pharm。罪。 2009 , 44 , 987–993。 [谷歌学术] - Shen, R.; Liu, D.; Hou, C.; Liu, D.; Zhao, L.; Cheng, J.; Wang, D.; Bai, D. Protective effect of Potentilla anserina polysaccharide on cadmium-induced nephrotoxicity in vitro and in vivo. Food Funct. 2017, 8, 3636–3646. [Google Scholar] [CrossRef]
沉,R。刘,D.;侯,C.;刘,D.;赵L.;程,J.;王,D。 Bai,D.委陵菜多糖对镉诱导的体外和体内肾毒性的保护作用。食品功能。 2017 , 8 , 3636–3646。 [谷歌学术] [交叉引用] - Cheng, J.; Liu, D.; Zhao, L.; Zhao, Q.; Zhang, X.; Wang, B.; Bai, D. Potentilla anserine L. polysaccharide inhibits cadmium-induced neurotoxicity by attenuating autophagy. Neurochem. Int. 2021, 147, 105045. [Google Scholar] [CrossRef]
程,J.;刘,D.;赵L.;赵,Q。张X;王,B.; Bai,D.委陵菜多糖通过减弱自噬抑制镉诱导的神经毒性。神经化学。国际。 2021 , 147 , 105045. [谷歌学术] [交叉引用] - Ide, M.; Sonoda, N.; Inoue, T.; Kimura, S.; Minami, Y.; Makimura, H.; Hayashida, E.; Hyodo, F.; Yamato, M.; Takayanagi, R. The dipeptidyl peptidase-4 inhibitor, linagliptin, improves cognitive impairment in streptozotocin-induced diabetic mice by inhibiting oxidative stress and microglial activation. PLoS ONE 2020, 15, e0228750. [Google Scholar] [CrossRef]
伊德,M.;索诺达,N.;井上,T.;木村,S.;南,Y.;牧村,H.;林田,E.;兵藤,F.;大和,M.; Takayanagi, R. 二肽基肽酶 4 抑制剂利格列汀通过抑制氧化应激和小胶质细胞活化来改善链脲佐菌素诱导的糖尿病小鼠的认知障碍。 PLoS ONE 2020,15 , e0228750。 [谷歌学术] [交叉引用] - Siddiqui, N.; Ali, J.; Parvez, S.; Zameer, S.; Najmi, A.K.; Akhtar, M. Linagliptin, a DPP-4 inhibitor, ameliorates Aβ (1− 42) peptides induced neurodegeneration and brain insulin resistance (BIR) via insulin receptor substrate-1 (IRS-1) in rat model of Alzheimer’s disease. Neuropharmacology 2021, 195, 108662. [Google Scholar] [CrossRef]
西迪基,N.;阿里,J.;帕尔韦兹,S.;扎梅尔,S.;阿拉斯加纳杰米; Akhtar, M. Linagliptin 是一种 DPP-4 抑制剂,可在阿尔茨海默病大鼠模型中通过胰岛素受体底物 1 (IRS-1) 改善 Aβ (1− 42) 肽诱导的神经变性和脑胰岛素抵抗 (BIR)。神经药理学2021 , 195 , 108662. [谷歌学术] [交叉引用] - Arab, H.H.; Eid, A.H.; Alsufyani, S.E.; Ashour, A.M.; El-Sheikh, A.A.K.; Darwish, H.W.; Georgy, G.S. Neuroprotective Impact of Linagliptin against Cadmium-Induced Cognitive Impairment and Neuropathological Aberrations: Targeting SIRT1/Nrf2 Axis, Apoptosis, and Autophagy. Pharmaceuticals 2023, 16, 1065. [Google Scholar] [CrossRef]
阿拉伯人,HH;开斋节,啊;阿尔苏菲亚尼,东南部;阿舒尔,上午;埃尔-谢赫,AAK;达尔维什,HW; Georgy,GS 利格列汀对镉引起的认知障碍和神经病理畸变的神经保护作用:针对 SIRT1/Nrf2 轴、细胞凋亡和自噬。制药2023 , 16 , 1065. [谷歌学术] [交叉引用] - Pagani, M.R.; Reisin, R.C.; Uchitel, O.D. Calcium Signaling Pathways Mediating Synaptic Potentiation Triggered by Amyotrophic Lateral Sclerosis IgG in Motor Nerve Terminals. J. Neurosci. 2006, 26, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
帕加尼先生;赖辛,RC; Uchitel,OD 钙信号通路介导运动神经末梢肌萎缩侧索硬化症 IgG 触发的突触增强。 J.神经科学。 2006 , 26 , 2661–2672。 [谷歌学术] [交叉引用] [ PubMed ] - Adeoye, T.; Shah, S.I.; Demuro, A.; Rabson, D.A.; Ullah, G. Upregulated Ca2+ Release from the Endoplasmic Reticulum Leads to Impaired Presynaptic Function in Familial Alzheimer’s Disease. Cells 2022, 11, 2167. [Google Scholar] [CrossRef] [PubMed]
阿德奥耶,T.;沙阿,SI;德穆罗,A.;拉布森,DA; Ullah, G. 内质网 Ca 2+释放上调导致家族性阿尔茨海默氏病突触前功能受损。细胞2022、11、2167 。 [谷歌学术][交叉引用][ PubMed ] - Tsentsevitsky, A.N.; Zakyrjanova, G.F.; Petrov, A.M. Cadmium desynchronizes neurotransmitter release in the neuromuscular junction: Key role of ROS. Free Radic. Biol. Med. 2020, 155, 19–28. [Google Scholar] [CrossRef] [PubMed]
岑采维茨基,AN;扎基尔贾诺娃,GF; Petrov,AM 镉使神经肌肉接头中的神经递质释放不同步:ROS 的关键作用。自由基。生物。医学。 2020 , 155 , 19–28。 [谷歌学术] [交叉引用] [ PubMed ] - Kaeser, P.S.; Regehr, W.G. Molecular Mechanisms for Synchronous, Asynchronous, and Spontaneous Neurotransmitter Release. Annu. Rev. Physiol. 2014, 76, 333. [Google Scholar] [CrossRef]
凯撒,PS; Regehr,同步、异步和自发神经递质释放的工作组分子机制。安努。生理学家牧师。 2014 , 76 , 333. [谷歌学术] [交叉引用] - Moriyama, Y.; Maeda, M.; Futai, M. The role of V-ATPase in neuronal and endocrine systems. J. Exp. Biol. 1992, 172, 171–178. [Google Scholar] [CrossRef]
森山,Y.;前田,M.; Futai, M. V-ATP 酶在神经元和内分泌系统中的作用。 J.Exp。生物。 1992 , 172 , 171–178。 [谷歌学术] [交叉引用] - Kosmidis, E.; Shuttle, C.G.; Preobraschenski, J.; Ganzella, M.; Johnson, P.J.; Veshaguri, S.; Holmkvist, J.; Møller, M.P.; Marantos, O.; Marcoline, F.; et al. Regulation of the mammalian-brain V-ATPase through ultraslow mode-switching. Nature 2022, 611, 827. [Google Scholar] [CrossRef]
科斯米迪斯,E.;航天飞机,重心;普雷奥布拉申斯基,J.;甘泽拉,M.;约翰逊,PJ;维沙古里,S.;霍姆奎斯特,J.;默勒,议员;马兰托斯,O.;马可琳,F.;等人。通过超慢模式切换调节哺乳动物大脑 V-ATP 酶。自然2022 , 611 , 827. [谷歌学术] [交叉引用] - Borisova, T.; Krisanova, N.; Sivko, R.; Kasatkina, L.; Borysov, A.; Griffin, S.; Wireman, M. Presynaptic malfunction: The neurotoxic effects of cadmium and lead on the proton gradient of synaptic vesicles and glutamate transport. Neurochem. Int. 2011, 59, 272–279. [Google Scholar] [CrossRef]
鲍里索娃,T.;克里萨诺瓦,N.;西夫科,R.;卡萨特金纳,L.;鲍里索夫,A.;格里芬,S.; Wireman, M. 突触前功能障碍:镉和铅对突触小泡质子梯度和谷氨酸转运的神经毒性作用。神经化学。国际。 2011 , 59 , 272–279。 [谷歌学术] [交叉引用] - Moyano, P.; de Frias, M.; Lobo, M.; Anadon, M.J.; Sola, E.; Pelayo, A.; Díaz, M.J.; Frejo, M.T.; Del Pino, J. Cadmium induced ROS alters M1 and M3 receptors, leading to SN56 cholinergic neuronal loss, through AChE variants disruption. Toxicology 2018, 394, 54–62. [Google Scholar] [CrossRef]
莫亚诺,P.;德弗里亚斯,M.;洛博,M.;阿纳东,MJ;索拉,E.;佩拉约,A.;迪亚兹,MJ;蒙大拿州弗雷霍; Del Pino, J. 镉诱导的 ROS 通过 AChE 变异破坏改变 M1 和 M3 受体,导致 SN56 胆碱能神经元损失。毒理学2018 , 394 , 54–62。 [谷歌学术] [交叉引用] - Lee, S.R. Critical Role of Zinc as Either an Antioxidant or a Prooxidant in Cellular Systems. Oxidative Med. Cell. Longev. 2018, 2018, 9156285-11. [Google Scholar] [CrossRef]
Lee,SR 锌在细胞系统中作为抗氧化剂或促氧化剂的关键作用。氧化医学。细胞。长寿。 2018 , 2018 , 9156285-11。 [谷歌学术] [交叉引用] - Garfinkel, D. Is aging inevitable?: The intracellular zinc deficiency hypothesis of aging. Med. Hypotheses 1986, 19, 117–137. [Google Scholar] [CrossRef]
加芬克尔,D。衰老是不可避免的吗?:衰老的细胞内锌缺乏假说。医学。假设1986 , 19 , 117–137。 [谷歌学术] [交叉引用] - Hong Bin, Q.; Garfinkel, D. The cadmium toxicity hypothesis of aging: A possible explanation for the zinc deficiency hypothesis of aging. Med. Hypotheses 1994, 42, 380–384. [Google Scholar]
洪斌,Q。 Garfinkel, D. 衰老的镉毒性假说:衰老的缺锌假说的可能解释。医学。假设1994 , 42 , 380–384。 [谷歌学术] - Paoletti, P.; Vergnano, A.M.; Barbour, B.; Casado, M. Zinc at glutamatergic synapses. Neuroscience 2009, 158, 126–136. [Google Scholar] [CrossRef] [PubMed]
保莱蒂,P.;韦尔尼亚诺,AM;巴伯,B.;卡萨多,M. 谷氨酸能突触的锌。神经科学2009 , 158 , 126–136。 [谷歌学术] [交叉引用] [ PubMed ] - Beyer, N.; Coulson, D.T.R.; Heggarty, S.; Ravid, R.; Brent Irvine, G.; Hellemans, J.; Johnston, J.A. ZnT3 mRNA levels are reduced in Alzheimer’s disease post-mortem brain|Molecular Neurodegeneration|Full Text. Mol. Neurodegener. 2009, 4, 53. [Google Scholar] [CrossRef] [PubMed]
拜尔,N.;科尔森,D.T.R.;赫加蒂,S.;拉维德,R.;布伦特·欧文,G.;海勒曼斯,J.; Johnston, JA 阿尔茨海默病死后大脑中 ZnT3 mRNA 水平降低|分子神经退行性变|全文。摩尔。神经退行性疾病。 2009 , 4 , 53. [谷歌学术][交叉引用][ PubMed ] - Adlard, P.A.; Parncutt, J.M.; Finkelstein, D.I.; Bush, A.I. Cognitive loss in zinc transporter-3 knock-out mice: A phenocopy for the synaptic and memory deficits of Alzheimer’s disease? J. Neurosci. 2010, 30, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
阿德拉德,宾夕法尼亚州;帕恩卡特,JM;芬克尔斯坦,DI; Bush, AI 锌转运蛋白 3 敲除小鼠的认知丧失:阿尔茨海默病突触和记忆缺陷的表型? J.神经科学。 2010 , 30 , 1631–1636。 [谷歌学术] [交叉引用] [ PubMed ] - Hajji, N.; Calvert, C.; Ritchie, C.W.; Sastre, M. The Role of Metals in Alzheimer’s Disease. Mech. Met. Involv. Neurodegener. Dis. 2013, 80. [Google Scholar] [CrossRef]
哈吉,N.;卡尔弗特,C.;里奇,C.W.; Sastre, M。金属在阿尔茨海默病中的作用。机甲。的。参与。神经退行性疾病。迪斯。 2013年, 80 。 [谷歌学术] [交叉引用] - Cuajungco, M.P.; Lees, G.J. Zinc and Alzheimer’s disease: Is there a direct link? Brain Res. Rev. 1997, 23, 219. [Google Scholar] [CrossRef]
Cuajungco,国会议员; Lees、GJ 锌和阿尔茨海默病:有直接联系吗?脑研究。修订版1997 , 23 , 219. [谷歌学术] [交叉引用] - Atrián-Blasco, E.; Conte-Daban, A.; Hureau, C. Mutual interference of Cu and Zn ions in Alzheimer’s disease: Perspectives at the molecular level. Dalton Trans. 2017, 46, 1275–12759. [Google Scholar] [CrossRef]
阿特里安-布拉斯科,E.;孔特-达班,A.; Hureau, C. 阿尔茨海默病中铜离子和锌离子的相互干扰:分子水平的视角。道尔顿跨。 2017 , 46 , 1275–12759。 [谷歌学术] [交叉引用] - Yuan, Y.; Niu, F.; Liu, Y.; Lu, N. Zinc and its effects on oxidative stress in Alzheimer’s disease. Neurol. Sci. 2014, 35, 923–928. [Google Scholar] [CrossRef]
袁,Y。牛,F。刘,Y。 Lu, N. 锌及其对阿尔茨海默病氧化应激的影响。内罗尔。科学。 2014 , 35 , 923–928。 [谷歌学术] [交叉引用] - Lavanya, R.D.; Reddy, B.S.; Abdul Sattar, S.; Rao, A.D.P. Trace element imbalances in blood serum of Alzheimer’s disease patients. Spectrosc. Lett. 2021, 54, 458–471. [Google Scholar] [CrossRef]
拉瓦尼亚,RD;雷迪,学士;阿卜杜勒·萨塔尔,S.; Rao,ADP 阿尔茨海默病患者血清中的微量元素失衡。光谱。莱特。 2021 , 54 , 458–471。 [谷歌学术] [交叉引用] - Li, K.; Li, A.; Mei, Y.; Zhao, J.; Zhou, Q.; Li, Y.; Yang, M.; Xu, Q. Trace elements and Alzheimer dementia in population-based studies: A bibliometric and meta-analysis. Environ. Pollut. 2023, 318, 120782. [Google Scholar] [CrossRef]
李,K。李,A。梅,Y。赵,J。周Q。李,Y。杨,M。 Xu, Q. 基于人群的研究中的微量元素和阿尔茨海默氏痴呆:文献计量和荟萃分析。环境。污染。 2023 , 318 , 120782. [谷歌学术] [交叉引用] - Xu, L.; Zhang, W.; Liu, X.; Zhang, C.; Wang, P.; Zhao, X. Circulatory Levels of Toxic Metals (Aluminum, Cadmium, Mercury, Lead) in Patients with Alzheimer’s Disease: A Quantitative Meta-Analysis and Systematic Review. J. Alzheimer’s Dis. 2018, 62, 361–372. [Google Scholar] [CrossRef] [PubMed]
徐L.;张,W。刘X。张,C.;王,P。赵,X。阿尔茨海默病患者中有毒金属(铝、镉、汞、铅)的循环水平:定量荟萃分析和系统评价。 J.阿尔茨海默病。 2018 , 62 , 361–372。 [谷歌学术] [交叉引用] [ PubMed ] - Jin, W. Regulation of BDNF-TrkB Signaling and Potential Therapeutic Strategies for Parkinson’s Disease. J. Clin. Med. 2020, 9, 257. [Google Scholar] [CrossRef] [PubMed]
Jin, W. BDNF-TrkB 信号传导的调节和帕金森病的潜在治疗策略。 J.克林。医学。 2020 , 9 , 257. [谷歌学术][交叉引用][ PubMed ] - Syme, C.D.; Nadal, R.C.; Rigby, S.E.J.; John, H. Viles Copper Binding to the Amyloid-beta (abeta) Peptide Associated with Alzheimer’s Disease. J. Biol. Chem. 2004, 279, 18169. [Google Scholar] [CrossRef] [PubMed]
赛姆,CD;纳达尔,RC;里格比,SEJ; John, H. Viles 铜与与阿尔茨海默氏病相关的淀粉样β (abeta) 肽的结合。 J.Biol。化学。 2004 , 279 , 18169. [谷歌学术] [ CrossRef ] [ PubMed ] - Boopathi, S.; Kolandaivel, P. Fe2+ binding on amyloid [beta]-peptide promotes aggregation. Proteins Struct. Funct. Bioinform. 2016, 84, 1257. [Google Scholar] [CrossRef]
布帕蒂,S.; Kolandaivel,P. Fe 2+与淀粉样β-肽的结合促进聚集。蛋白质结构。功能。生物信息。 2016 , 84 , 1257. [谷歌学术] [交叉引用] - Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 1045, 13, 1045. [Google Scholar] [CrossRef]
沃德,RJ;祖卡,FA;杜恩,JH;克莱顿,RR; Zecca, L. 铁在大脑衰老和神经退行性疾病中的作用。柳叶刀神经醇。 1045、13、1045 。[谷歌学术][交叉引用] - Bauer, H.; Krizbai, I.A.; Bauer, H.; Traweger, A. “You Shall Not Pass”–tight junctions of the blood brain barrier. Front. Neurosci. 2014, 8, 392. [Google Scholar] [CrossRef]
鲍尔,H.;爱荷华州克里兹拜;鲍尔,H.; Traweger, A.“你不能通过”——血脑屏障的紧密连接。正面。神经科学。 2014 , 8 , 392. [谷歌学术] [交叉引用] - Luissint, A.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P. Tight junctions at the blood brain barrier: Physiological architecture and disease-associated dysregulation|Fluids and Barriers of the CNS|Full Text. Fluids Barriers CNS 2012, 9, 23. [Google Scholar] [CrossRef]
路易斯特,A.;阿图斯,C.;冰川,F.;加尼沙莫西,K.; Couraud, P. 血脑屏障的紧密连接:生理结构和疾病相关的失调|中枢神经系统的液体和屏障|全文。流体屏障 CNS 2012 , 9 , 23. [ Google Scholar ] [ CrossRef ] - Wang, L.; Xiong, X.; Zhang, L.; Shen, J. Neurovascular Unit: A critical role in ischemic stroke. CNS Neurosci. Ther. 2021, 27, 7–16. [Google Scholar] [CrossRef]
王L.;熊X。张L。 Shen, J. 神经血管单元:在缺血性中风中的关键作用。中枢神经系统神经科学。瑟尔。 2021 年, 27 日, 7-16 日。 [谷歌学术] [交叉引用] - Mendez-Armenta, M.; Rios, C. Cadmium neurotoxicity. Environ. Toxicol. Pharmacol. 2007, 23, 350–358. [Google Scholar] [CrossRef]
门德斯-阿门塔,M.; Rios, C. 镉的神经毒性。环境。毒性。药理学。 2007 , 23 , 350–358。 [谷歌学术] [交叉引用] - Rai, A.; Maurya, S.K.; Khare, P.; Srivastava, A.; Bandyopadhyay, S. Characterization of Developmental Neurotoxicity of As, Cd, and Pb Mixture: Synergistic Action of Metal Mixture in Glial and Neuronal Functions. Toxicol. Sci. 2010, 118, 586–601. [Google Scholar] [CrossRef]
拉伊,A.;孔雀,SK;卡雷,P.;斯里瓦斯塔瓦,A.; Bandyopadhyay, S. As、Cd 和 Pb 混合物的发育神经毒性的表征:金属混合物在神经胶质和神经元功能中的协同作用。毒性。科学。 2010 , 118 , 586–601。 [谷歌学术] [交叉引用] - Carrino, D.; Branca, J.J.V.; Becatti, M.; Paternostro, F.; Morucci, G.; Gulisano, M.; Di Cesare Mannelli, L.; Pacini, A. Alcohol-Induced Blood-Brain Barrier Impairment: An In Vitro Study. Int. J. Environ. Res. Public Health 2021, 18, 2683. [Google Scholar] [CrossRef] [PubMed]
卡里诺,D.;布兰卡,JJV;贝卡蒂,M.;帕特诺斯特罗,F.;莫鲁奇,G.;古利萨诺,M.;迪·切萨雷·曼内利,L.; Pacini, A. 酒精引起的血脑屏障损伤:一项体外研究。国际。 J.环境。资源。公共卫生2021 , 18 , 2683. [谷歌学术] [ CrossRef ] [ PubMed ] - Pal, R.; Nath, R.; Dipgill, K. Influence of ethanol on cadmium accumulation and its impact on lipid peroxidation and membrane bound functional enzymes (Na+, K+-ATPASE and acetylcholinesterase) in various regions of adult rat brain. Neurochem. Int. 1993, 23, 451. [Google Scholar] [CrossRef] [PubMed]
帕尔,R。内斯,R。 Dipgill, K. 乙醇对成年大鼠脑各区域镉积累的影响及其对脂质过氧化和膜结合功能酶(Na + 、K + -ATP酶和乙酰胆碱酯酶)的影响。神经化学。国际。 1993 , 23 , 451. [谷歌学术] [交叉引用] [ PubMed ] - Zhang, T.; Xu, Z.; Wen, L.; Lei, D.; Li, S.; Wang, J.; Huang, J.; Wang, N.; Durkan, C.; Liao, X.; et al. Cadmium-induced dysfunction of the blood-brain barrier depends on ROS-mediated inhibition of PTPase activity in zebrafish. J. Hazard. Mater. 2021, 412, 125198. [Google Scholar] [CrossRef] [PubMed]
张,T。徐,Z。文L.;雷,D。李,S。王,J。黄,J。王,N。杜尔坎,C.;廖X.;等人。镉引起的血脑屏障功能障碍取决于 ROS 介导的斑马鱼 PTP 酶活性抑制。 J·阿扎尔。马特。 2021 , 412 , 125198. [谷歌学术][交叉引用][ PubMed ] - Branca, J.J.V.; Maresca, M.; Morucci, G.; Mello, T.; Becatti, M.; Pazzagli, L.; Colzi, I.; Gonnelli, C.; Carrino, D.; Paternostro, F.; et al. Effects of Cadmium on ZO-1 Tight Junction Integrity of the Blood Brain Barrier. Int. J. Mol. Sci. 2019, 20, 6010. [Google Scholar] [CrossRef] [PubMed]
布兰卡,JJV;马雷斯卡,M.;莫鲁奇,G.;梅洛,T.;贝卡蒂,M.;帕扎利,L.;科尔齐,I.;冈内利,C.;卡里诺,D.;帕特诺斯特罗,F.;等人。镉对血脑屏障 ZO-1 紧密连接完整性的影响。国际。 J.莫尔。科学。 2019 , 20 , 6010. [谷歌学术] [ CrossRef ] [ PubMed ] - Lohmann, C.; Krischke, M.; Wegener, J.; Galla, H. Tyrosine phosphatase inhibition induces loss of blood–brain barrier integrity by matrix metalloproteinase-dependent and -independent pathways. Brain Res. 2004, 995, 184–196. [Google Scholar] [CrossRef]
洛曼,C.;克里施克,M.;韦格纳,J.; Galla, H. 酪氨酸磷酸酶抑制通过基质金属蛋白酶依赖性和非依赖性途径诱导血脑屏障完整性丧失。脑研究, 2004,995,184-196 。 [谷歌学术] [交叉引用] - Kim, S.; Cheon, H.; Kim, S.; Kim, Y. GSK-3β-mediated regulation of cadmium-induced cell death and survival. Cell Mol. Biol. Lett. 2018, 23, 9. [Google Scholar] [CrossRef]
金,S。 Cheon,H.;金,S。 Kim, Y. GSK-3β 介导的镉诱导细胞死亡和存活的调节。细胞分子。生物。莱特。 2018 , 23 , 9. [谷歌学术] [交叉引用] - Maier, J.; Locatelli, L.; Fedele, G.; Cazzaniga, A.; Mazur, A. Magnesium and the Brain: A Focus on Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2022, 24, 223. [Google Scholar] [CrossRef]
迈尔,J.;洛卡特利,L.;费德勒,G.;卡扎尼加,A.; Mazur, A. 镁与大脑:关注神经炎症和神经变性。国际。 J.莫尔。科学。 2022 , 24 , 223. [谷歌学术] [交叉引用] - Streit, W.J.; Mrak, R.E.; Griffin, W.S.T. Microglia and neuroinflammation: A pathological perspective|Journal of Neuroinflammation|Full Text. J. Neuroinflamm. 2004, 1, 14. [Google Scholar] [CrossRef]
斯特雷特,WJ;穆拉克,RE; Griffin,WST 小胶质细胞和神经炎症:病理学视角|神经炎症杂志|全文。 J.神经炎症。 2004 , 1 , 14. [谷歌学术] [交叉引用] - DiSabato, D.; Quan, N.; Godbout, J.P. Neuroinflammation: The Devil Is in the Details. J. Neurochem. 2016, 139, 136–153. [Google Scholar] [CrossRef]
迪萨巴托,D.;泉,N.; Godbout,JP 神经炎症:细节决定成败。 J.神经化学。 2016 , 139 , 136–153。 [谷歌学术] [交叉引用] - Gilhus, N.E.; Deuschl, G. Neuroinflammation—A common thread in neurological disorders. Nat. Rev. Neurol. 2019, 15, 429–430. [Google Scholar] [CrossRef]
内布拉斯加州吉尔胡斯; Deuschl, G. 神经炎症——神经系统疾病的常见线索。纳特。尼罗尔牧师。 2019 , 15 , 429–430。 [谷歌学术] [交叉引用] - Wells, E.; Hacohen, Y.; Waldman, A.; Tillema, J.M.; Soldatos, A.; Ances, B.; Benseler, S.; Bielekova, B.; Dale, R.C.; Dalmau, J.; et al. Neuroimmune disorders of the central nervous system in children in the molecular era. Nat. Rev. Neurol. 2018, 14, 433–445. [Google Scholar] [CrossRef] [PubMed]
威尔斯,E.;哈科恩,Y.;沃尔德曼,A.;蒂勒玛,JM;索尔达托斯,A.;安塞斯,B.;本塞尔,S.;别莱科娃,B.;戴尔,RC;达尔毛,J.;等人。分子时代儿童中枢神经系统的神经免疫疾病。纳特。尼罗尔牧师。 2018 , 14 , 433–445。 [谷歌学术] [交叉引用] [ PubMed ] - Martínez-Hernández, M.I.; Acosta-Saavedra, L.C.; Hernández-Kelly, L.C.; Loaeza-Loaeza, J.; Ortega, A. Microglial Activation in Metal Neurotoxicity: Impact in Neurodegenerative Diseases. Biomed. Res. Int. 2023, 2023, 7389508. [Google Scholar] [CrossRef] [PubMed]
马丁内斯-埃尔南德斯,密歇根州;阿科斯塔-萨维德拉,LC;埃尔南德斯-凯利,LC;洛埃萨-洛埃萨,J.; Ortega, A. 金属神经毒性中的小胶质细胞激活:对神经退行性疾病的影响。生物医学。资源。国际。 2023 , 2023 , 7389508.[谷歌学术][交叉引用][ PubMed ] - Yang, Z.; Yang, S.; Qian, S.Y.; Hong, J.; Kadiiska, M.B.; Tennant, R.W.; Waalkes, M.P.; Liu, J. Cadmium-Induced Toxicity in Rat Primary Mid-brain Neuroglia Cultures: Role of Oxidative Stress from Microglia. Toxicol. Sci. 2007, 98, 488–494. [Google Scholar] [CrossRef]
杨,Z。杨,S。钱 SY;洪,J。卡迪斯卡,MB;田南特,RW;沃尔克斯,议员; Liu, J. 大鼠原代中脑神经胶质细胞培养物中镉诱导的毒性:小胶质细胞氧化应激的作用。毒性。科学。 2007 , 98 , 488–494。 [谷歌学术] [交叉引用] - Khan, A.; Ikram, M.; Muhammad, T.; Park, J.; Kim, M.O. Caffeine Modulates Cadmium-Induced Oxidative Stress, Neuroinflammation, and Cognitive Impairments by Regulating Nrf-2/HO-1 In Vivo and In Vitro. J. Clin. Med. 2019, 8, 680. [Google Scholar] [CrossRef]
汗,A.;伊克拉姆,M.;穆罕默德,T.;帕克,J.; Kim,MO 咖啡因通过在体内和体外调节 Nrf-2/HO-1 来调节镉引起的氧化应激、神经炎症和认知障碍。 J.克林。医学。 2019 , 8 , 680. [谷歌学术] [交叉引用] - Górska, A.; Markiewicz-Gospodarek, A.; Markiewicz, R.; Chilimoniuk, Z.; Borowski, B.; Trubalski, M.; Czarnek, K. Distribution of Iron, Copper, Zinc and Cadmium in Glia, Their Influence on Glial Cells and Relationship with Neurodegenerative Diseases. Brain Sci. 2023, 13, 911. [Google Scholar] [CrossRef] [PubMed]
戈尔斯卡,A.;马基维奇-戈斯波达雷克,A.;马凯维奇,R.;奇利莫尼克,Z.;博罗夫斯基,B.;特鲁巴尔斯基,M.; Czarnek, K. 神经胶质细胞中铁、铜、锌和镉的分布、它们对神经胶质细胞的影响以及与神经退行性疾病的关系。脑科学。 2023 , 13 , 911. [谷歌学术] [ CrossRef ] [ PubMed ] - Raichle, M.E.; Posner, J.B.; Plum, F. Cerebral Blood Flow During. Arch. Neurol. 1970, 23, 394–403. [Google Scholar] [CrossRef]
雷切尔,缅因州;波斯纳,JB; Plum, F. 期间的脑血流。拱。内罗尔。 1970 , 23 , 394–403。 [谷歌学术] [交叉引用] - Mathieu, C.; de la Sierra-Gallay, I.L.; Duval, R.; Xu, X.; Cocaign, A.; Léger, T.; Woffendin, G.; Camadro, J.; Etchebest, C.; Haouz, A.; et al. Insights into Brain Glycogen Metabolism: The Structure of Human Brain Glycogen Phosphorylase. J. Biol. Chem. 2016, 291, 18072–18083. [Google Scholar] [CrossRef]
马蒂厄,C.;伊利诺伊州德拉塞拉-加莱;杜瓦尔,R。徐,X。科卡恩,A.;莱热,T.;沃芬丁,G.;卡马德罗,J.;埃切贝斯特,C.;豪兹,A.;等人。深入了解脑糖原代谢:人脑糖原磷酸化酶的结构。 J.Biol。化学。 2016 , 291 , 18072–18083。 [谷歌学术] [交叉引用] - Wang, Y.; Ma, K.; Wang, P.; Baba, O.; Zhang, H.; Parent, J.M.; Zheng, P.; Liu, Y.; Minassian, B.A.; Liu, Y. Laforin Prevents Stress-Induced Polyglucosan Body Formation and Lafora Disease Progression in Neurons. Mol. Neurobiol. 2013, 48, 49–61. [Google Scholar] [CrossRef]
王,Y。马,K。王,P。巴巴,O.;张,H。家长,JM;郑,P。刘,Y。米纳西安,学士; Liu, Y. Laforin 可防止神经元中应激诱导的聚葡萄糖体形成和 Lafora 疾病进展。摩尔。神经生物学。 2013 , 48 , 49–61。 [谷歌学术] [交叉引用] - Saez, I.; Duran, J.; Sinadinos, C.; Beltran, A.; Yanes, O.; María, F.T.; Carlos, M.-P.; Marco, M.; Joan, J.G. Neurons Have an Active Glycogen Metabolism that Contributes to Tolerance to Hypoxia. J. Cereb. Blood Flow Metab. 2014, 34, 945–955. [Google Scholar] [CrossRef]
赛斯,I。杜兰,J.;西纳迪诺斯,C.;贝尔特兰,A.;亚内斯,O.;玛丽亚,金融时报;卡洛斯,M.-P.;马可,M.; Joan,JG 神经元具有活跃的糖原代谢,有助于耐受缺氧。 J·塞雷布。血流代谢。 2014 , 34 , 945–955。 [谷歌学术] [交叉引用] - Tischner, K. Cadmium. In Experimental and Clinical Neurotoxicology, 1st ed.; Oxford Univeristy Press: Oxford, UK, 1980; pp. 345–355. [Google Scholar]
Tischner, K. 镉。实验和临床神经毒理学,第一版;牛津大学出版社:英国牛津,1980 年;第 345–355 页。 [谷歌学术] - Mathieu, C.; Dupret, J.; Rodrigues-Lima, F. The Structure and the Regulation of Glycogen Phosphorylases in Brain. In Brain Glycogen Metabolism; DiNuzzo, M., Schousboe, A., Eds.; Springer International Publishing: Cham, Switzerland, 2019; pp. 125–145. [Google Scholar]
马蒂厄,C.;杜普雷,J.;罗德里格斯-利马,F。脑中糖原磷酸化酶的结构和调节。在脑糖原代谢中; DiNuzzo,M.,Schousboe,A.,编辑;施普林格国际出版社:Cham,瑞士,2019 年;第 125–145 页。 [谷歌学术] - Rai, A.; Singh, P.K.; Singh, V.; Kumar, V.; Mishra, R.; Thakur, A.K.; Mahadevan, A.; Shankar, S.K.; Jana, N.R.; Ganesh, S. Glycogen synthase protects neurons from cytotoxicity of mutant huntingtin by enhancing the autophagy flux. Cell Death Dis. 2018, 9, 201. [Google Scholar] [CrossRef]
拉伊,A.;辛格,PK;辛格,V.;库马尔,V.;米什拉,R.;阿拉斯加塔库尔;马哈德万,A.;香卡,SK;贾纳,NR; Ganesh, S. 糖原合酶通过增强自噬通量来保护神经元免受突变亨廷顿蛋白的细胞毒性。细胞死亡疾病。 2018 , 9 , 201. [谷歌学术] [交叉引用] - Smith, E.E.; Taylor, P.M.; Whelan, W.J. Enzymic Processes in Glycogen Metabolism. In Carbohydrate Metabolism and Its Disorders; Dickens, F., Whelan, W.J., Randale, P.J., Eds.; Academic Press: London, UK; New York, NY, USA, 1968; Volume I, pp. 89–133. [Google Scholar]
史密斯,EE;泰勒,PM; Whelan, WJ 糖原代谢中的酶过程。碳水化合物代谢及其紊乱;狄更斯,F.,惠兰,WJ,兰德尔,PJ,编辑;学术出版社:英国伦敦;美国纽约州纽约市,1968 年;第一卷,第 89–133 页。 [谷歌学术] - Lew, C.R.; Guin, S.; Theodorescu, D. Targeting glycogen metabolism in bladder cancer. Nat. Rev. Urol. 2015, 12, 383–391. [Google Scholar] [CrossRef] [PubMed]
卢,CR;吉恩,S.; Theodorescu, D. 针对膀胱癌的糖原代谢。夜晚。牧师。乌罗尔。 2015,12,383-391 。 [谷歌学术] [交叉引用] [ PubMed ] - Kielan, Z.; Ziółkowska, B.; Falkus, B.; Jethon, Z. Effect of cadmium intoxication on glucose utilization in energy metabolism of muscles. Acta Physiol. Pol. 1989, 40, 535–543. [Google Scholar] [PubMed]
基兰,Z.;齐奥科斯卡,B.;法尔克斯,B.;杰森,Z。镉中毒对肌肉能量代谢中葡萄糖利用的影响。生理学报。波尔。 1989 , 40 , 535–543。 [谷歌学术] [考研] - Hazelhoff Roelfzema, W.; Hacker, H.J.; Van Noorden, C.J.F. Effects of Cadmium Exposure on Glycogen Phosphorylase Activity in Rat Placenta as Demonstrated by Histochemical Means. Histochemistry 1989, 91, 305–308. [Google Scholar] [CrossRef]
哈泽尔霍夫·罗尔夫泽马,W.;黑客,HJ; Van Noorden,CJF 通过组织化学方法证明镉暴露对大鼠胎盘糖原磷酸化酶活性的影响。组织化学1989 , 91 , 305–308。 [谷歌学术] [交叉引用] - Miseta, A.; Csutora, P. Relationship Between the Occurrence of Cysteine in Proteins and the Complexity of Organisms. Mol. Biol. Evol. 2000, 17, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
米塞塔,A.; Csutora, P. 蛋白质中半胱氨酸的出现与生物体复杂性之间的关系。摩尔。生物。进化。 2000 , 17 , 1232–1239。 [谷歌学术] [交叉引用] [ PubMed ] - Smith, C.; Dicaire, M.; Brais, B.; La Piana, R. Neurological Involvement in Glycogen Storage Disease Type IXa due to PHKA2 Mutation. Can. J. Neurol. Sci. 2020, 47, 400–403. [Google Scholar] [CrossRef]
史密斯,C.;迪凯尔,M.;布雷斯,B.; La Piana, R. PHKA2 突变导致 IXa 型糖原累积病的神经学参与。能。 J.内罗尔。科学。 2020 , 47 , 400–403。 [谷歌学术] [交叉引用] - Muzetti, J.H.; do Valle, D.A.; Santos, M.L.S.F.; Telles, B.A.; Cordeiro, M.L. Neurological Characteristics of Pediatric Glycogen Storage Disease. Front. Endocrinol. 2021, 12, 685272. [Google Scholar] [CrossRef]
穆泽蒂,JH;多瓦莱,DA;桑托斯,MLSF;泰勒斯,文学学士; Cordeiro,ML 小儿糖原累积病的神经学特征。正面。内分泌。 2021 , 12 , 685272. [谷歌学术] [交叉引用] - Canibano-Fraile, R.; Harlaar, L.; dos Santos, C.A.; Hoogeveen-Westerveld, M.; Demmers, J.A.A.; Snijders, T.; Lijnzaad, P.; Verdijk, R.M.; van der Beek Nadine, A.M.E.; van Doorn, P.A.; et al. Lysosomal glycogen accumulation in Pompe disease results in disturbed cytoplasmic glycogen metabolism. J. Inherit. Metab. Dis. 2023, 46, 101–115. [Google Scholar] [CrossRef] [PubMed]
卡尼巴诺-弗莱尔,R.;哈拉尔,L.;加利福尼亚州多斯桑托斯;霍赫芬-韦斯特维尔德,M.;德默斯,JAA;斯奈德斯,T.;利恩扎德,P.;维迪克,RM;范德贝克纳丁,AME;宾夕法尼亚州范多恩;等人。庞贝病中的溶酶体糖原积累导致细胞质糖原代谢紊乱。 J.继承。代谢物。迪斯。 2023 , 46 , 101–115。 [谷歌学术] [交叉引用] [ PubMed ] - Taylor, K.M.; Meyers, E.; Phipps, M.; Kishnani, P.S.; Cheng, S.H.; Scheule, R.K.; Moreland, R.J. Dysregulation of Multiple Facets of Glycogen Metabolism in a Murine Model of Pompe Disease. PLoS ONE 2013, 8, e56181. [Google Scholar] [CrossRef] [PubMed]
泰勒,KM;迈耶斯,E.;菲普斯,M.;基什纳尼,PS;程SH;斯科勒,RK; Moreland,RJ 庞贝病小鼠模型中糖原代谢多个方面的失调。 《公共图书馆一号》 2013 年, 8 ,e56181。 [谷歌学术] [交叉引用] [ PubMed ] - Khanh, D.N.N.; Vy, N.T.T.; Phuong, T.H.; Nhi, P.T.; Thang, N.Q.; Sy, D.T.; Phuong, N.T.K. Effects of Cadmium and Lead on Muscle and Liver Glycogen Levels of Climbing Perch (Anabas testudineus). Bull. Environ. Contam. Toxicol. 2022, 108, 854–860. [Google Scholar] [CrossRef]
Khanh,DNN;维,NTT;芳,TH;尼,PT;唐,NQ;西,DT; Phuong,NTK 镉和铅对攀鲈( Anabas testudineus )肌肉和肝糖原水平的影响。公牛。环境。康塔姆。毒性。 2022 , 108 , 854–860。 [谷歌学术] [交叉引用] - Dwivedi, K. International Journal of Advanced Research in Biological Sciences Effect of Cadmium on Liver Glycogen Reserve and its Size in Albino Rats. Int. J. Adv. Res. Biol. Sci. 2021, 8, 150–154. [Google Scholar]
Dwivedi, K. 国际生物科学高级研究杂志镉对白化大鼠肝糖原储备及其大小的影响。国际。 J.Adv。资源。生物。科学。 2021 , 8 , 150–154。 [谷歌学术] - Buha, A.; Đukić-Ćosić, D.; Ćurčić, M.; Bulat, Z.; Antonijević, B.; Moulis, J.; Goumenou, M.; Wallace, D. Emerging Links between Cadmium Exposure and Insulin Resistance: Human, Animal, and Cell Study Data. Toxics 2020, 8, 63. [Google Scholar] [CrossRef] [PubMed]
布哈,A.;杜基奇-乔西奇,D.;丘尔西奇,M.;布拉特,Z.;安东尼耶维奇,B.;穆里斯,J.;古梅努,M.; Wallace, D. 镉暴露与胰岛素抵抗之间的新兴联系:人类、动物和细胞研究数据。有毒物质2020 , 8 , 63. [ Google Scholar ] [ CrossRef ] [ PubMed ] - Fang, X.; Yu, S.X.; Lu, Y.; Bast, R.C.; Woodgett, J.R.; Mills, G.B. Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc. Natl. Acad. Sci. USA 2000, 97, 11960–11965. [Google Scholar] [CrossRef] [PubMed]
方X.;余SX;卢,Y。韧皮,RC;伍德吉特,JR; Mills,GB 蛋白激酶 A 对糖原合酶激酶 3 的磷酸化和失活。Proc 。国家。阿卡德。科学。美国2000 , 97 , 11960–11965。 [谷歌学术] [交叉引用] [ PubMed ] - Shati, A.A.; Alfaifi, M.Y. Trans-resveratrol Inhibits Tau Phosphorylation in the Brains of Control and Cadmium Chloride-Treated Rats by Activating PP2A and PI3K/Akt Induced-Inhibition of GSK3β. Neurochem. Res. 2019, 44, 357–373. [Google Scholar] [CrossRef] [PubMed]
沙蒂,AA; Alfaifi,MY 反式白藜芦醇通过激活 PP2A 和 PI3K/Akt 诱导的 GSK3β 抑制,抑制对照大鼠和氯化镉处理大鼠大脑中的 Tau 磷酸化。神经化学。资源。 2019 , 44 , 357–373。 [谷歌学术] [交叉引用] [ PubMed ] - De Zwaan, A.; Zandee, D.I. The utilization of glycogen and accumulation of some intermediates during anaerobiosis in Mytilus edulis L. Comp. Biochem. Physiol. Part B Comp. Biochem. 1972, 43, 47–54. [Google Scholar] [CrossRef]
德兹万,A.; Zandee,DI贻贝厌氧过程中糖原的利用和一些中间体的积累。生物化学。生理学。 B 部分比较。生物化学。 1972 , 43 , 47–54。 [谷歌学术] [交叉引用] - Lee, B.; Kim, Y. Association of Blood Cadmium Level with Metabolic Syndrome After Adjustment for Confounding by Serum Ferritin and Other Factors: 2008–2012 Korean National Health and Nutrition Examination Survey. Biol. Trace Elem. Res. 2016, 171, 6–16. [Google Scholar] [CrossRef]
李,B.; Kim, Y. 调整血清铁蛋白和其他因素混杂后的血镉水平与代谢综合征的关联:2008-2012 年韩国国家健康和营养检查调查。生物。跟踪埃莱姆。资源。 2016,171,6-16 。 [谷歌学术] [交叉引用] - Filippini, T.; Wise, L.A.; Vinceti, M. Cadmium exposure and risk of diabetes and prediabetes: A systematic review and dose-response meta-analysis. Environ. Int. 2022, 158, 106920. [Google Scholar] [CrossRef]
菲利皮尼,T.;怀斯,洛杉矶; Vinceti, M. 镉暴露与糖尿病和糖尿病前期的风险:系统评价和剂量反应荟萃分析。环境。国际。 2022 , 158 , 106920. [谷歌学术] [交叉引用] - Gong, P.; Chang, X.; Chen, X.; Bai, X.; Wen, H.; Pi, S.; Yang, W.; Wang, L.; Chen, F. Metabolomics study of cadmium-induced diabetic nephropathy and protective effect of caffeic acid phenethyl ester using UPLC-Q-TOF-MS combined with pattern recognition. Environ. Toxicol. Pharmacol. 2017, 54, 80–92. [Google Scholar] [CrossRef]
龚,P。张,X。陈X.;白,X。温,H。皮,S。杨,W。王L.; Chen,F。利用 UPLC-Q-TOF-MS 结合模式识别对镉诱导的糖尿病肾病的代谢组学研究以及咖啡酸苯乙酯的保护作用。环境。毒性。药理学。 2017 , 54 , 80–92。 [谷歌学术] [交叉引用] - Bak, L.K.; Walls, A.B.; Schousboe, A.; Waagepetersen, H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018, 293, 7108–7116. [Google Scholar] [CrossRef]
巴克,LK;沃尔斯,AB;斯科斯博,A.; Waagepetersen,HS 健康和患病大脑中的星形胶质细胞糖原代谢。 J.Biol。化学。 2018 , 293 , 7108–7116。 [谷歌学术] [交叉引用] - Islam, F.; Shohag, S.; Akhter, S.; Islam, M.R.; Sultana, S.; Mitra, S.; Chandran, D.; Khandaker, M.U.; Ashraf, G.M.; Idris, A.M.; et al. Exposure of metal toxicity in Alzheimer’s disease: An extensive review. Front. Pharmacol. 2022, 13, 903099. [Google Scholar] [CrossRef]
伊斯兰,F.;肖哈格,S.;阿赫特,S.;伊斯兰先生;苏丹娜,S.;米特拉,S.;钱德兰,D.;坎达克,MU;阿什拉夫,总经理;伊德里斯,AM;等人。阿尔茨海默病中金属毒性的暴露:广泛的回顾。正面。药理学。 2022 , 13 , 903099. [谷歌学术] [交叉引用] - Duran, J.; Gruart, A.; García-Rocha, M.; Delgado-García, J.M.; Guinovart, J.J. Glycogen accumulation underlies neurodegeneration and autophagy impairment in Lafora disease. Hum. Mol. Genet. 2014, 23, 3147–3156. [Google Scholar] [CrossRef]
杜兰,J.;格鲁特,A.;加西亚-罗查,M.;德尔加多-加西亚,JM; Guinovart,JJ 糖原积累是拉福拉病神经变性和自噬损伤的基础。哼。摩尔。热内特. 2014 , 23 , 3147–3156。 [谷歌学术] [交叉引用] - Huat, T.J.; Camats-Perna, J.; Newcombe, E.A.; Valmas, N.; Kitazawa, M.; Medeiros, R. Metal Toxicity Links to Alzheimer’s Disease and Neuroinflammation. J. Mol. Biol. 2019, 431, 1843–1868. [Google Scholar] [CrossRef] [PubMed]
发,TJ;卡马茨-佩纳,J.;纽科姆 (EA);瓦尔马斯,N.;北泽,M.; Medeiros, R. 金属毒性与阿尔茨海默病和神经炎症有关。 J.莫尔。生物。 2019 , 431 , 1843–1868。 [谷歌学术] [交叉引用] [ PubMed ] - Lavoie, S.; Allaman, I.; Petit, J.; Do, K.Q.; Magistretti, P.J. Altered Glycogen Metabolism in Cultured Astrocytes from Mice with Chronic Glutathione Deficit; Relevance for Neuroenergetics in Schizophrenia. PLoS ONE 2011, 6, e22875. [Google Scholar] [CrossRef] [PubMed]
拉沃伊,S.;阿拉曼,I。佩蒂特,J.;做,KQ; Magistretti,PJ 改变了慢性谷胱甘肽缺乏小鼠培养的星形胶质细胞中的糖原代谢;精神分裂症神经能量学的相关性。 《公共图书馆一号》 2011 年, 6 ,e22875。 [谷歌学术] [交叉引用] [ PubMed ] - Wang, J.; Zhu, H.; Wang, K.; Yang, Z.; Liu, Z. Protective effect of quercetin on rat testes against cadmium toxicity by alleviating oxidative stress and autophagy. Environ. Sci. Pollut. Res. 2020, 27, 25278–25286. [Google Scholar] [CrossRef] [PubMed]
王,J。朱,H.;王,K。杨,Z。 Liu,Z。槲皮素通过减轻氧化应激和自噬对大鼠睾丸抵抗镉毒性的保护作用。环境。科学。污染。资源。 2020 , 27 , 25278–25286。 [谷歌学术] [交叉引用] [ PubMed ] - Alshammari, G.M.; Al-Qahtani, W.H.; Alshuniaber, M.A.; Yagoub, A.E.A.; Al-Khalifah, A.S.; Al-Harbi, L.N.; Alhussain, M.H.; AlSedairy, S.A.; Yahya, M.A. Quercetin improves the impairment in memory function and attenuates hippocampal damage in cadmium chloride-intoxicated male rats by suppressing acetylcholinesterase and concomitant activation of SIRT1 signaling. J. Funct. Foods 2021, 86, 104675. [Google Scholar] [CrossRef]
阿尔沙马里,总经理;卡塔尼,WH;马萨诸塞州阿尔舒尼亚伯;亚古布,AEA;阿勒哈利法,AS;路易斯安那州阿尔哈比;阿尔侯赛因,MH; AlSedairy,SA; Yahya, MA 槲皮素通过抑制乙酰胆碱酯酶并同时激活 SIRT1 信号传导,改善氯化镉中毒雄性大鼠的记忆功能损伤并减轻海马损伤。 J.函数。食品2021 , 86 , 104675。 [谷歌学术] [交叉引用] - Kini, R.D.; Arunkumar, N.; Gokul, M. Potential Protective Role of Beta Carotene on Cadmium Induced Brain and Kidney Damage. Indian J. Public Health Res. Dev. 2019, 10, 532–535. [Google Scholar] [CrossRef]
基尼,RD;阿伦库马尔,N.; Gokul, M. β-胡萝卜素对镉引起的脑和肾损伤的潜在保护作用。印度 J. 公共卫生研究中心。开发。 2019 , 10 , 532–535。 [谷歌学术] [交叉引用] - Wiberg, E.; Wiberg, N. Inorganic Chemistry; Academic Press: Cambridge, MA, USA, 2001. [Google Scholar]
维伯格,E.;维伯格,N.无机化学;学术出版社:美国马萨诸塞州剑桥,2001 年。[谷歌学术] - Cranton, E.M.; Zheng, X.L.; Smith, I.M. Urinary trace and toxic elements and minerals in untimed urine specimens relative to urine creatinine. J. Adv. Med. 1989, 1, 331–397. [Google Scholar]
克兰顿,EM;郑XL; Smith,IM 不定期尿液样本中相对于尿肌酸酐的尿液痕量以及有毒元素和矿物质。 J.Adv。医学。 1989,1,331-397 。 [谷歌学术] - Waters, R.S.; Bryden, N.A.; Patterson, K.Y.; Veillon, C.; Anderson, R.A. EDTA chelation effects on urinary losses of cadmium, calcium, chromium, cobalt, copper, lead, magnesium, and zinc. Biol. Trace Elem. Res. 2001, 83, 207–221. [Google Scholar] [CrossRef]
沃特斯,RS;北卡罗来纳州布赖登;肯塔基州帕特森;韦永,C.; Anderson,RA EDTA 螯合对镉、钙、铬、钴、铜、铅、镁和锌尿损失的影响。生物。跟踪埃莱姆。资源。 2001 , 83 , 207–221。 [谷歌学术] [交叉引用] - Fulgenzi, A.; Vietti, D.; Ferrero, M.E. EDTA Chelation Therapy in the Treatment of Neurodegenerative Diseases: An Update. Biomedicines 2020, 8, 269. [Google Scholar] [CrossRef]
富尔根齐,A.;维埃蒂,D.; Ferrero,ME EDTA 螯合疗法治疗神经退行性疾病:更新。生物医学2020 , 8 , 269. [谷歌学术] [交叉引用] - Mostafa, D.G.; Khaleel, E.F.; Badi, R.M.; Abdel-Aleem, G.; Abdeen, H.M. Rutin hydrate inhibits apoptosis in the brains of cadmium chloride-treated rats via preserving the mitochondrial integrity and inhibiting endoplasmic reticulum stress. Neurol. Res. 2019, 41, 594–608. [Google Scholar] [CrossRef]
穆斯塔法,DG;卡雷尔,EF;巴迪,RM;阿卜杜勒-阿利姆,G.; Abdeen,HM 芦丁水合物通过保留线粒体完整性和抑制内质网应激来抑制氯化镉处理的大鼠大脑中的细胞凋亡。内罗尔。资源。 2019 , 41 , 594–608。 [谷歌学术] [交叉引用] - Bjørklund, G.; Crisponi, G.; Nurchi, V.M.; Cappai, R.; Buha Djordjevic, A.; Aaseth, J. A Review on Coordination Properties of Thiol-Containing Chelating Agents Towards Mercury, Cadmium, and Lead. Molecules 2019, 24, 3247. [Google Scholar] [CrossRef]
比约克伦德,G.;克里斯波尼,G.;努尔奇,VM;卡佩,R.;布哈·乔尔杰维奇,A.; Aaseth, J. 含硫醇螯合剂对汞、镉和铅的配位特性综述。分子2019 , 24 , 3247. [谷歌学术] [交叉引用] - Rahimzadeh, M.R.; Rahimzadeh, M.R.; Kazemi, S.; Moghadamnia, A.A. Review Article Cadmium toxicity and treatment: An update. Casp. J. Intern. Med. 2017, 8, 135. [Google Scholar]
拉希姆扎德先生;拉希姆扎德先生;卡泽米,S.; Moghadamnia,AA 评论文章镉毒性和治疗:更新。卡斯普。 J.实习生。医学。 2017 , 8 , 135. [谷歌学术] - Harris, C.J.; Voss, K.; Murchison, C.; Ralle, M.; Frahler, K.; Carter, R.; Rhoads, A.; Lind, B.; Robinson, E.; Quinn, J.F. Oral Zinc Reduces Amyloid Burden in Tg2576 Mice. J. Alzheimer’s Dis. 2014, 41, 179–192. [Google Scholar] [CrossRef] [PubMed]
哈里斯,CJ;沃斯,K.;默奇森,C.;拉勒,M.;弗拉勒,K.;卡特,R.;罗兹,A.;林德,B.;罗宾逊,E.; Quinn, JF 口服锌可减少 Tg2576 小鼠的淀粉样蛋白负担。 J.阿尔茨海默病。 2014 , 41 , 179–192。 [谷歌学术] [交叉引用] [ PubMed ]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. 免责声明/出版商注:所有出版物中包含的声明、意见和数据仅代表个人作者和贡献者的观点,而非 MDPI 和/或编辑的观点。 MDPI 和/或编辑对内容中提及的任何想法、方法、说明或产品造成的任何人员或财产伤害不承担任何责任。 |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
© 2023 作者版权所有。被许可方 MDPI,瑞士巴塞尔。本文是根据知识共享署名 (CC BY) 许可证 ( https://creativecommons.org/licenses/by/4.0/ ) 的条款和条件分发的开放获取文章。
Share and Cite 分享和引用
Arruebarrena, M.A.; Hawe, C.T.; Lee, Y.M.; Branco, R.C.
Mechanisms of Cadmium Neurotoxicity. Int. J. Mol. Sci. 2023, 24, 16558.
https://doi.org/10.3390/ijms242316558IF: 4.9 Q1 B2
马萨诸塞州阿鲁巴雷纳;康涅狄格州霍威;李,YM; Branco,RC 镉神经毒性机制。国际。 J.莫尔。科学。 2023 , 24 , 16558。https://doi.org/10.3390/ijms242316558如果:4.9 Q1 B2
Arruebarrena MA, Hawe CT, Lee YM, Branco RC.
Mechanisms of Cadmium Neurotoxicity. International Journal of Molecular Sciences. 2023; 24(23):16558.
https://doi.org/10.3390/ijms242316558IF: 4.9 Q1 B2
Arruebarrena MA、Hawe CT、Lee YM、Branco RC。镉神经毒性的机制。国际分子科学杂志。 2023; 24(23):16558。 https://doi.org/10.3390/ijms242316558如果:4.9 Q1 B2
Arruebarrena, Madelyn A., Calvin T. Hawe, Young Min Lee, and Rachel C. Branco.
2023. "Mechanisms of Cadmium Neurotoxicity" International Journal of Molecular Sciences 24, no. 23: 16558.
https://doi.org/10.3390/ijms242316558IF: 4.9 Q1 B2
Arruebarrena、Madelyn A.、Calvin T. Hawe、Young Min Lee 和 Rachel C. Branco。 2023.“镉神经毒性的机制”国际分子科学杂志24,第 1 期。 23:16558。https://doi.org/10.3390/ijms242316558如果:4.9 Q1 B2
请注意,从2016年第一期开始,该期刊使用文章编号而不是页码。请参阅此处的更多详细信息。

{kind=link}
{kind=link}
{kind=link}
{kind=link}